
Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
Arvelig nefritt (Alports syndrom) hos barn
Medisinsk ekspert av artikkelen

Sist anmeldt: 05.07.2025
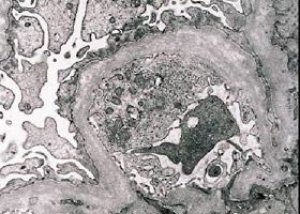
Arvelig nefritt (Alports syndrom) er en genetisk bestemt arvelig ikke-immun glomerulopati, manifestert av hematuri (noen ganger med proteinuri), progressiv nedgang i nyrefunksjon med utvikling av kronisk nyresvikt, ofte kombinert med sensorinevral døvhet og synshemming.
Sykdommen ble først beskrevet i 1902 av LG Guthrie, som observerte en familie der hematuri ble observert i flere generasjoner. I 1915 beskrev AF Hurst utviklingen av uremi hos medlemmer av samme familie. I 1927 identifiserte A. Alport først hørselstap hos flere slektninger med hematuri. På 1950-tallet ble øyelesjoner ved en lignende sykdom beskrevet. I 1972, hos pasienter med arvelig hematuri, under en morfologisk studie av nyrevev, avdekket Hinglais et al. ujevn ekspansjon og lagdeling av glomerulære basalmembraner. I 1985 ble det genetiske grunnlaget for arvelig nefritt identifisert - en mutasjon i type IV-kollagengenet (Fiengold et al., 1985).
Studien av sykdommens genetiske natur lot oss konkludere med at forskjellene i de fenotypiske manifestasjonene av arvelig nefritt (med eller uten hørselstap) skyldes graden av uttrykk av det mutante genet. Dermed anses for tiden alle kliniske varianter som manifestasjoner av én sykdom, og begrepet "arvelig nefritt" er synonymt med begrepet "Alports syndrom".
I følge epidemiologiske studier forekommer arvelig nefritt med en frekvens på 17 per 100 000 barn.
 [
[ Årsaker til Alports syndrom
Det genetiske grunnlaget for sykdommen er en mutasjon i genet til a-5-kjeden av type IV-kollagen. Denne typen er universell for basalmembranene i nyrene, cochlea-apparatet, linsekapselen, netthinnen og hornhinnen i øyet, noe som er bevist i studier med monoklonale antistoffer mot denne kollagenfraksjonen. Nylig har muligheten for å bruke DNA-prober for prenatal diagnostikk av arvelig nefritt blitt indikert.
Det understrekes viktigheten av å teste alle familiemedlemmer med DNA-prober for å identifisere bærere av det mutante genet, noe som er av stor betydning for å gjennomføre medisinsk og genetisk rådgivning av familier med denne sykdommen. Imidlertid har opptil 20 % av familiene ikke slektninger som lider av nyresykdom, noe som tyder på en høy frekvens av spontane mutasjoner av det unormale genet. De fleste pasienter med arvelig nefritt har individer med nyresykdom, hørselstap og synspatologi i familiene sine; blodsekteskap mellom personer med en eller flere forfedre er viktige, siden sannsynligheten for å motta de samme genene fra begge foreldrene øker i ekteskap med beslektede individer. Autosomalt dominante, autosomalt recessive og dominante, X-bundne overføringsveier er etablert.
Hos barn skilles det oftest mellom tre typer arvelig nefritt: Alports syndrom, arvelig nefritt uten hørselstap og familiær benign hematuri.
Alports syndrom er en arvelig nefritt med hørselshemming. Den er basert på en kombinert defekt i strukturen til kollagenet i den glomerulære basalmembranen i nyrene, øret og øyet. Genet for klassisk Alports syndrom er lokalisert i locus 21-22q på den lange armen av X-kromosomet. I de fleste tilfeller arves det på en dominant måte, knyttet til X-kromosomet. I denne forbindelse er Alports syndrom mer alvorlig hos menn, siden funksjonen til det mutante genet hos kvinner kompenseres av et sunt allel av det andre, uskadede kromosomet.
Det genetiske grunnlaget for utviklingen av arvelig nefritt er mutasjoner i genene til alfakjedene i type IV-kollagen. Seks alfakjeder av type IV-kollagen G er kjent: genene til a5- og a6-kjedene (Col4A5 og Col4A5) er lokalisert på den lange armen til X-kromosomet i 21-22q-sonen; genene til a3- og a4-kjedene (Col4A3 og Col4A4) er på det andre kromosomet; genene til a1- og a2-kjedene (Col4A1 og Col4A2) er på det 13. kromosomet.
I de fleste tilfeller (80–85 %) oppdages et X-bundet arvemønster av sykdommen, assosiert med skade på Col4A5-genet som følge av delesjon, punktmutasjoner eller skjøtingsforstyrrelser. For tiden er det funnet mer enn 200 mutasjoner av Col4A5-genet, som er ansvarlige for forstyrrelsen av syntesen av a5-kjedene av type IV-kollagen. Ved denne typen arv manifesterer sykdommen seg hos barn av begge kjønn, men hos gutter er den mer alvorlig.
Mutasjoner i lociene til Col4A3- og Col4A4-genene som er ansvarlige for syntesen av a3- og a4-kjedene av type IV-kollagen, arves autosomalt. Ifølge forskning observeres den autosomalt dominante arvetypen i 16 % av tilfellene av arvelig nefritt, og den autosomalt recessive typen observeres hos 6 % av pasientene. Omtrent 10 varianter av mutasjoner av Col4A3- og Col4A4-genene er kjent.
Resultatet av mutasjoner er et brudd på monteringsprosessene til type IV-kollagen, noe som fører til et brudd på strukturen. Type IV-kollagen er en av hovedkomponentene i den glomerulære basalmembranen, cochlea-apparatet og øyets linse, hvis patologi vil bli oppdaget i klinikken for arvelig nefritt.
Kollagen type IV, som er en del av den glomerulære basalmembranen, består hovedsakelig av to a1-kjeder (IV) og én a2-kjede (IV), og inneholder også a3-, a4- og a5-kjeder. Oftest, ved X-bundet arv, er mutasjonen av Col4A5-genet ledsaget av fravær av a3-, a4-, a5- og a6-kjeder i strukturen til kollagen type IV, og antallet o1- og a2-kjeder i den glomerulære basalmembranen øker. Mekanismen bak dette fenomenet er uklar, det antas at årsaken er post-transkripsjonelle endringer i mRNA.
Fraværet av a3-, a4- og a5-kjeder i strukturen til type IV-kollagen i glomerulære basalmembraner fører til tynning og skjørhet i de tidlige stadiene av Alports syndrom, som klinisk manifesteres oftere ved hematuri (sjeldnere ved hematuri med proteinuri eller bare proteinuri), hørselstap og lenticonus. Videre progresjon av sykdommen fører til fortykkelse og nedsatt permeabilitet av basalmembranene i de sene stadiene av sykdommen, med spredning av kollagen av typene V og VI i dem, manifestert i en økning i proteinuri og en reduksjon i nyrefunksjon.
Arten av mutasjonen som ligger til grunn for arvelig nefritt bestemmer i stor grad dens fenotypiske manifestasjon. Ved delesjon av X-kromosomet med samtidig mutasjon av Col4A5- og Col4A6-genene som er ansvarlige for syntesen av a5- og a6-kjedene av type IV-kollagen, er Alports syndrom kombinert med leiomyomatose i spiserøret og kjønnsorganene. Ifølge forskningsdata observeres en større alvorlighetsgrad av den patologiske prosessen ved en mutasjon av Col4A5-genet assosiert med en delesjon, en kombinasjon av nyreskade med ekstrarenale manifestasjoner og tidlig utvikling av kronisk nyresvikt, sammenlignet med en punktmutasjon av dette genet.
Morfologisk sett avslører elektronmikroskopi fortynning og lagdeling av glomerulære basalmembraner (spesielt lamina densa) og tilstedeværelse av elektrontette granuler. Glomerulære lesjoner kan være heterogene hos samme pasient, fra minimale fokale mesangiale lesjoner til glomerulosklerose. Glomerulitt ved Alports syndrom er alltid immunnegativ, noe som skiller den fra glomerulonefritt. Karakteristiske trekk inkluderer utvikling av tubulær atrofi, lymfohistiocytisk infiltrasjon og tilstedeværelse av "skumceller" med lipidinneslutninger - lipofager. Etter hvert som sykdommen utvikler seg, avsløres fortykkelse og uttalt ødeleggelse av glomerulære basalmembraner.
Visse endringer i immunsystemet avdekkes. Pasienter med arvelig nefritt har et redusert IgA-nivå og en tendens til å øke IgM-konsentrasjonen i blodet. IgG-nivået kan være forhøyet i de tidlige stadiene av sykdommen og synke i de senere stadiene. Kanskje er økningen i IgM- og G-konsentrasjon en slags kompenserende reaksjon som respons på IgA-mangel.
Den funksjonelle aktiviteten til T-lymfocyttsystemet reduseres; en selektiv reduksjon i B-lymfocytter som er ansvarlige for syntesen av Ig A observeres, den fagocytiske koblingen til immunitet forstyrres, hovedsakelig på grunn av forstyrrelse av kjemotaksi og intracellulære fordøyelsesprosesser i nøytrofiler.
Ved undersøkelse av nyrebiopsi hos pasienter med Alports syndrom avslører elektronmikroskopidata ultrastrukturelle endringer i glomerulære basalmembraner: tynning, strukturforstyrrelse og splitting av glomerulære basalmembraner med endring i tykkelse og ujevne konturer. I de tidlige stadiene av arvelig nefritt bestemmer defekten tynningen og skjørheten til glomerulære basalmembraner.
Tynning av glomerulære membraner er et gunstigere tegn og er vanligere hos jenter. Et mer konstant elektronmikroskopisk tegn ved arvelig nefritt er splitting av basalmembranen, og alvorlighetsgraden av ødeleggelsen korrelerer med alvorlighetsgraden av prosessen.
Symptomer på Alports syndrom hos barn
De første symptomene på Alports syndrom i form av isolert urinsyndrom oppdages oftest hos barn i de første tre leveårene. I de fleste tilfeller oppdages sykdommen tilfeldig. Urinsyndrom oppdages under en forebyggende undersøkelse av barnet, før innleggelse i barnehage eller under ARVI. Ved patologi i urinen under ARVI. Ved arvelig nefritt, i motsetning til ervervet glomerulonefritt, er det ingen latent periode.
I den innledende fasen av sykdommen lider barnets helse lite, et karakteristisk trekk er vedvarende og motstandsdyktig urinveissyndrom. Et av hovedtegnene er hematuri av varierende alvorlighetsgrad, observert i 100 % av tilfellene. En økning i graden av hematuri observeres under eller etter luftveisinfeksjoner, fysisk aktivitet eller etter forebyggende vaksinasjoner. Proteinuri overstiger i de fleste tilfeller ikke 1 g/dag, i begynnelsen av sykdommen kan være ustabil, etter hvert som prosessen utvikler seg, øker proteinuri. Med jevne mellomrom kan leukocyturi med en overvekt av lymfocytter være tilstede i urinsedimentet, noe som er assosiert med utviklingen av interstitielle forandringer.
Deretter svekkes delvis nyrefunksjon, pasientens allmenntilstand forverres: rus, muskelsvakhet, arteriell hypotensjon, ofte hørselshemming (spesielt hos gutter), og noen ganger synshemming. Rus manifesterer seg ved blekhet, tretthet og hodepine. I den første fasen av sykdommen oppdages hørselstap i de fleste tilfeller kun ved audiografi. Hørselstap ved Alports syndrom kan oppstå i forskjellige perioder av barndommen, men oftest diagnostiseres hørselstap i alderen 6-10 år. Hørselstap hos barn begynner med høye frekvenser, når en betydelig grad i luft- og benledning, og går over fra lydledende til lydoppfattende hørselstap. Hørselstap kan være et av de første symptomene på sykdommen og kan gå forut for urinveissyndrom.
I 20 % av tilfellene har pasienter med Alports syndrom forandringer i synsorganene. De hyppigst oppdagede anomaliene er linsens: sfærofoki, fremre, bakre eller blandet lenticonus, og ulike typer grå stær. I familier med Alports syndrom er det en betydelig forekomst av nærsynthet. En rekke forskere bemerker stadig bilaterale perimakulære forandringer i disse familiene i form av lyse hvitaktige eller gulaktige granulasjoner i corpus luteum. De anser dette tegnet som et konstant symptom som har høy diagnostisk verdi ved Alports syndrom. KS Chugh et al. (1993) fant i en oftalmologisk studie hos pasienter med Alports syndrom en reduksjon i synsskarphet i 66,7 % av tilfellene, fremre lenticonus i 37,8 %, retinale flekker i 22,2 %, grå stær i 20 % og keratokonus i 6,7 %.
Hos noen barn med arvelig nefritt, spesielt når nyresvikt utvikler seg, observeres en betydelig forsinkelse i fysisk utvikling. Etter hvert som nyresvikten utvikler seg, utvikler arteriell hypertensjon seg. Hos barn oppdages det oftere i ungdomsårene og i eldre aldersgrupper.
Pasienter med arvelig nefritt kjennetegnes av tilstedeværelsen av forskjellige (mer enn 5-7) stigmaer av bindevevsdysmorfogenese. Blant bindevevsstigmaene hos pasienter er de vanligste hypertelorisme i øynene, høy gane, bittanomalier, unormal form på ørene, krumning av lillefingeren på hendene og "sandalgap" på føttene. Arvelig nefritt kjennetegnes av ensartetheten av dysmorfogenese-stigmaer innenfor en familie, samt en høy frekvens av deres fordeling blant slektninger til probander langs hvis linje sykdommen overføres.
I de tidlige stadiene av sykdommen oppdages en isolert reduksjon i partielle nyrefunksjoner: transport av aminosyrer, elektrolytter, konsentrasjonsfunksjon, syreogenese, senere endringer påvirker funksjonstilstanden til både den proksimale og distale delen av nefronet og er preget av kombinerte partielle lidelser. En reduksjon i glomerulær filtrasjon forekommer senere, oftere i ungdomsårene. Etter hvert som arvelig nefritt utvikler seg, utvikles anemi.
Dermed er arvelig nefritt karakterisert ved et trinnvis sykdomsforløp: først et latent stadium eller skjulte kliniske symptomer, manifestert av minimale endringer i urinsyndromet, deretter skjer en gradvis dekompensasjon av prosessen med en reduksjon i nyrefunksjon med manifeste kliniske symptomer (forgiftning, asteni, utviklingsforsinkelse, anemi). Kliniske symptomer oppstår vanligvis uavhengig av lagdelingen av den inflammatoriske reaksjonen.
Arvelig nefritt kan manifestere seg i ulike aldersperioder, noe som avhenger av genets virkning, som er i en undertrykt tilstand inntil en viss tid.
Klassifikasjon
Det finnes tre typer arvelig nefritt
- Alternativ I - manifesterer seg klinisk som nefritt med hematuri, hørselstap og øyeskade. Nefrittforløpet er progressivt med utvikling av kronisk nyresvikt. Arvetypen er dominant, knyttet til X-kromosomet. Morfologisk avsløres et brudd på basalmembranens struktur, dens tynning og splitting.
- Alternativ II – manifesterer seg klinisk som nefritt med hematuri uten hørselstap. Nefrittforløpet er progressivt med utvikling av kronisk nyresvikt. Arvetypen er dominant, knyttet til X-kromosomet. Morfologisk påvises fortynning av den glomerulære kapillærbasalmembranen (spesielt laminadensa).
- Alternativ III - benign familiær hematuri. Forløpet er gunstig, kronisk nyresvikt utvikler seg ikke. Arvetypen er autosomal dominant eller autosomal recessiv. Ved autosomal recessive arvetyper observeres et mer alvorlig sykdomsforløp hos kvinner.
Diagnose av Alports syndrom
Følgende kriterier foreslås:
- tilstedeværelsen av minst to pasienter med nefropati i hver familie;
- hematuri som det ledende symptomet på nefropati hos probanden;
- tilstedeværelsen av hørselstap hos minst ett familiemedlem;
- utvikling av kronisk nyresvikt hos en eller flere slektninger.
I diagnostikk av ulike arvelige og medfødte sykdommer gis det stor plass til en helhetlig tilnærming til undersøkelse, og fremfor alt å ta hensyn til dataene som er innhentet ved sammenstilling av barnets stamtavle. Diagnosen Alport syndrom anses som gyldig i tilfeller der 3 av 4 typiske tegn oppdages hos pasienten: tilstedeværelse av hematuri og kronisk nyresvikt i familien, tilstedeværelse av nevrosensorisk hørselstap, synspatologi hos pasienten, påvisning av tegn på spalting av glomerulær basalmembran med endring i tykkelse og ujevne konturer under elektronmikroskopiske karakteristikker av biopsien.
Pasientundersøkelsen bør omfatte kliniske og genetiske forskningsmetoder; målrettet studie av sykdomshistorie; generell undersøkelse av pasienten under hensyntagen til diagnostisk signifikante kriterier. I kompensasjonsstadiet kan patologi kun oppdages ved å fokusere på slike syndromer som tilstedeværelsen av en arvelig byrde, hypotensjon, flere stigmaer av dysembryogenese, endringer i urinsyndromet. I dekompensasjonsstadiet kan ekstrarenale symptomer oppstå, som alvorlig rus, asteni, forsinket fysisk utvikling, anemi, som manifesterer seg og intensiveres med en gradvis reduksjon i nyrefunksjonen. Hos de fleste pasienter, med en reduksjon i nyrefunksjon, observeres følgende: redusert acido- og aminogenese; 50 % av pasientene merker en betydelig reduksjon i nyrenes sekretoriske funksjon; begrenset utvalg av svingninger i urinens optiske tetthet; forstyrrelse av filtreringsrytmen, og deretter en reduksjon i glomerulær filtrasjon. Stadiet med kronisk nyresvikt diagnostiseres når pasienter har et forhøyet nivå av urea i blodserumet (mer enn 0,35 g/l) i 3–6 måneder eller mer, og en reduksjon i glomerulær filtrasjon til 25 % av normen.
Differensialdiagnostikk av arvelig nefritt bør primært utføres ved den hematuriske formen av ervervet glomerulonefritt. Ervervet glomerulonefritt har oftest en akutt debut, en periode på 2-3 uker etter en infeksjon, ekstrarenale tegn, inkludert hypertensjon fra de første dagene (ved arvelig nefritt, tvert imot hypotensjon), redusert glomerulær filtrasjon ved sykdomsdebut, ingen svekkelse av partielle tubulære funksjoner, mens de ved arvelig er tilstede. Ervervet glomerulonefritt forekommer med mer uttalt hematuri og proteinuri, med økt ESR. Typiske endringer i glomerulære basalmembran, karakteristisk for arvelig nefritt, er av diagnostisk verdi.
Differensialdiagnostikk fra dysmetabolisk nefropati utføres ved kronisk nyresvikt, i familien klinisk påviste heterogene nyresykdommer, og det kan være et spekter av nefropati fra pyelonefritt til urolithiasis. Barn har ofte klager over smerter i magen og periodisk under vannlating, i urinsedimentet - oksalater.
Ved mistanke om arvelig nefritt bør pasienten henvises til en spesialisert nefrologisk avdeling for å avklare diagnosen.
Hva trenger å undersøke?
Hvordan undersøke?
Hvilke tester er nødvendig?
Hvem skal kontakte?
Behandling av Alports syndrom
Kuren inkluderer restriksjoner på tung fysisk anstrengelse og eksponering for frisk luft. Kostholdet er komplett, med tilstrekkelige nivåer av komplette proteiner, fett og karbohydrater, med tanke på nyrefunksjonen. Av stor betydning er deteksjon og behandling av kroniske infeksjonsfokus. Følgende medisiner brukes: ATP, kokarboksylase, pyridoksin (opptil 50 mg/dag), karnitinklorid. Kurene administreres 2-3 ganger i året. Ved hematuri foreskrives urtemedisin - brennesle, aroniajuice, ryllik.
Det finnes rapporter i utenlandsk og innenlandsk litteratur om behandling med prednisolon og bruk av cytostatika. Det er imidlertid vanskelig å bedømme effekten.
Ved kronisk nyresvikt brukes hemodialyse og nyretransplantasjon.
Det finnes ingen metoder for spesifikk (effektiv patogenetisk) terapi for arvelig nefritt. Alle behandlingstiltak er rettet mot å forhindre og bremse nedgangen i nyrefunksjonen.
Kostholdet bør være balansert og kaloririkt, med tanke på nyrenes funksjonelle tilstand. Ved fravær av funksjonsforstyrrelser bør barnets kosthold inneholde tilstrekkelig med proteiner, fett og karbohydrater. Ved tegn på nyresvikt bør mengden protein, karbohydrater, kalsium og fosfor begrenses, noe som forsinker utviklingen av kronisk nyresvikt.
Fysisk aktivitet bør begrenses; barn anbefales å unngå sport.
Kontakt med smittsomme pasienter bør unngås, og risikoen for å utvikle akutte luftveissykdommer bør reduseres. Sanering av foci med kronisk infeksjon er nødvendig. Forebyggende vaksinasjoner utføres ikke for barn med arvelig nefritt, vaksinasjon er kun mulig ved epidemiologiske indikasjoner.
Hormonell og immunsuppressiv behandling ved arvelig nefritt er ineffektiv. Det finnes indikasjoner på en viss positiv effekt (reduksjon i proteinuri og bremsing av sykdomsprogresjon) ved langvarig flerårig bruk av ciklosporin A og ACE-hemmere.
Ved behandling av pasienter brukes legemidler som forbedrer stoffskiftet:
- pyridoksin - 2–3 mg/kg/dag i 3 doser i 4 uker;
- kokarboksylase - 50 mg intramuskulært annenhver dag, totalt 10-15 injeksjoner;
- ATP - 1 ml intramuskulært annenhver dag, 10-15 injeksjoner;
- vitamin A - 1000 IE/år/dag i 1 dose i 2 uker;
- Vitamin E - 1 mg/kg/dag i 1 dose i 2 uker.
Denne typen terapi bidrar til å forbedre pasientenes generelle tilstand, redusere tubulære dysfunksjoner og utføres i kurer 3 ganger i året.
Levamisol kan brukes som en immunmodulator - 2 mg/kg/dag 2-3 ganger i uken med pauser mellom dosene på 3-4 dager.
Ifølge forskningsdata har hyperbarisk oksygenering en positiv effekt på alvorlighetsgraden av hematuri og nyredysfunksjon.
Den mest effektive metoden for behandling av arvelig nefritt er rettidig nyretransplantasjon. I dette tilfellet er det ikke tilbakefall av sykdommen ved transplantasjon; i en liten prosentandel av tilfellene (ca. 5 %) kan nefritt utvikles i den transplanterte nyren assosiert med antigener til glomerulære basalmembran.
En lovende retning er prenatal diagnostikk og genteknologisk terapi. Dyreforsøk viser høy effektivitet ved overføring av normale gener som er ansvarlige for syntesen av type IV kollagen-alfakjeder til nyrevevet, hvoretter syntesen av normale kollagenstrukturer observeres.
Prognose
Prognosen for arvelig nefritt er alltid alvorlig.
Prognostisk ugunstige kriterier for forløpet av arvelig nefritt er:
- mannlig kjønn;
- tidlig utvikling av kronisk nyresvikt hos familiemedlemmer;
- proteinuri (mer enn 1 g/dag);
- fortykkelse av glomerulære basalmembraner i henhold til mikroskopi;
- akustisk nevritt;
- delesjon i Col4A5-genet.
Prognosen for benign familiær hematuri er gunstigere.