
Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
Lissencefali i hjernen
Medisinsk ekspert av artikkelen

Sist anmeldt: 04.07.2025
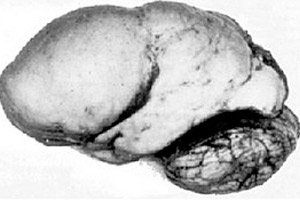
Blant organiske cerebrale patologier skiller en slik medfødt anomali i hjerneutviklingen som lissencefali seg ut, hvis essens ligger i den nesten glatte overflaten av hjernebarken på dens hemisfærer - med et utilstrekkelig antall viklinger og furer. [ 1 ]
Ved fullstendig fravær av konvolusjoner defineres agyri, og tilstedeværelsen av flere brede, flate konvolusjoner kalles pachygyri. Disse defektene, som noen andre reduksjonsdeformasjoner i hjernen, har koden Q04.3 i ICD-10.
Epidemiologi
Ifølge statistikk over sjeldne sykdommer er det 1–1,2 tilfeller av lissencefali per 100 000 nyfødte. [ 2 ], [ 3 ]
Ifølge noen data observeres opptil 25–30 % av tilfellene av klassisk lissencefali hos barn med Miller-Dieker syndrom; punktmutasjoner og delesjoner av LIS1- og DCX-genene oppdages hos nesten 85 % av pasientene. [ 4 ]
Genetiske studier av 17 gener assosiert med lissensfali viste at LIS1-mutasjon eller -delesjon står for 40 % av pasientene, og 23 % er assosiert med DCX-mutasjon, etterfulgt av TUBA1A (5 %) og DYNC1H1 (3 %).[ 5 ]
Fører til lissencefali
Alle kjente årsaker til dannelsen av hjernebarken (cortex cerebri) nesten eller helt uten foldinger og furer, som øker hjernens "arbeidsområde" og sikrer sentralnervesystemets "produktivitet", er forbundet med forstyrrelser i dens perinatale utvikling. Det vil si at lissensfali utvikler seg hos fosteret. [ 6 ]
Manglende dannelse av lag i hjernebarken til fosteret ved lissencefali er et resultat av unormal migrasjon av nevronene som danner den eller for tidlig opphør av denne prosessen.
Denne prosessen, som er essensiell for cerebrokortikal histogenese, skjer i flere stadier fra 7. til 18. svangerskapsuke. Og gitt den økte følsomheten for genetiske mutasjoner, samt ulike negative fysiske, kjemiske og biologiske påvirkninger, kan ethvert avvik fra normen føre til feil lokalisering av nevroner med mulig dannelse av et fortykket lag med grå substans i cortex uten en karakteristisk struktur. [ 7 ]
I noen tilfeller er lissensfali hos barn assosiert med Miller-Dieker-, Walker-Warburg- eller Norman-Roberts-syndromer.
Les også – Utviklingsdefekter i hjernen
Risikofaktorer
I tillegg til mutasjoner i noen gener, inkluderer risikofaktorer for fødselen av et barn med en så alvorlig defekt oksygenmangel (hypoksi) hos fosteret; utilstrekkelig blodtilførsel til hjernen (hypoperfusjon); akutt cerebrovaskulær hendelse i form av perinatalt hjerneslag; placentapatologi; virusinfeksjoner hos den gravide kvinnen (inkludert TORCH); [ 8 ] problemer med generell metabolisme og skjoldbruskkjertelfunksjon; røyking, alkohol, psykotrope og narkotiske stoffer; bruk av en rekke medisiner; økte strålingsnivåer. [ 9 ]
Patogenesen
Ikke alle tilfeller av lissensfali har en patogenese forårsaket av kromosomavvik og genmutasjoner. Men noen gener er kjent som koder for proteiner som spiller en viktig rolle i riktig bevegelse av nevroblaster og nevroner langs de radiale gliacellene – for å danne hjernebarken. Og mutasjoner av disse genene fører til denne patologien. [ 10 ]
Dette er spesielt sporadiske mutasjoner (uten arvelighet) av LIS1-genet på kromosom 17, som regulerer det cytoplasmatiske motorproteinet i mikrotubuli dynein, samt DCX-genet på X-kromosomet, som koder for proteinet doublecortin (lissencephalin-X). [ 11 ]. I det første tilfellet definerer spesialister klassisk lissencefali (type I), i det andre – X-bundet. [ 12 ]
Når FLN1-genet, som koder for fosfoproteinet filamin 1, deleteres, kan det hende at prosessen med styrt nevronmigrasjon ikke starter i det hele tatt, noe som fører til et fullstendig fravær av konvolusjoner (agyri). [ 13 ]
Mutasjoner er identifisert i CDK5-genet, som koder for et kinaseenzym – en katalysator for intracellulær metabolisme, som regulerer cellesyklusen i CNS-nevroner og sikrer deres normale migrasjon under den prenatale dannelsen av hjernestrukturer.
Unormale endringer i RELN-genet på kromosom 7 som forårsaker kortikale gyrale defekter ved Norman-Roberts syndrom resulterer i en mangel på det ekstracellulære glykoproteinet reelin, som er nødvendig for å regulere migrasjonen og plasseringen av nevrale stamceller under utviklingen av hjernebarken. [ 14 ], [ 15 ], [ 16 ]
ARX-genet koder for ikke-aristalens homeobox-protein, en transkripsjonsfaktor som spiller en viktig rolle i forhjernen og andre vev.[ 17 ] Barn med ARX-mutasjonen har andre symptomer, som manglende deler av hjernen (agenese av corpus callosum), unormale kjønnsorganer og alvorlig epilepsi.[ 18 ],[ 19 ]
Flere gener har blitt knyttet til lissensfali. Disse genene inkluderer VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A og CDK5.[ 20 ]
Cytomegalovirus (CMV) har vært assosiert med utvikling av lissencefali på grunn av redusert blodtilførsel til fosterhjernen. Alvorlighetsgraden av CMV-infeksjon avhenger av svangerskapsalderen. Tidlig infeksjon er mer sannsynlig å forårsake lissencefali fordi neuronal migrasjon skjer tidlig i svangerskapet.[ 21 ]
I tillegg inkluderer mekanismen for forekomst av denne anomalien ufullstendig eller senere opphør av migrasjon av nevroner fra den periventrikulære generative sonen til hjernebarken. Og i slike tilfeller utvikles enten ufullstendig lissensfali eller pachygyri, der flere brede riller og konvolusjoner dannes (men de fleste av dem er fraværende).
Symptomer lissencefali
De første tegnene på denne patologien (i fravær av de tidligere nevnte syndromene) kan ikke vises umiddelbart etter fødselen, men etter halvannen til to måneder. Og oftest observeres følgende kliniske symptomer på lissensfali:
- muskelhypotoni, ofte kombinert med spastisk lammelse;
- kramper og generaliserte tonisk-kloniske anfall (i form av opisthotonus);
- alvorlig psykisk utviklingshemming og veksthemming;
- svekkelse av nevrologiske og motoriske funksjoner.
Problemer med å svelge forårsaker vanskeligheter med å mate babyen. [ 22 ]
En høy grad av nevromotorisk svekkelse manifesterer seg ofte som tetraplegi – lammelse av alle lemmer. Deformasjon av hender, fingre eller tær er mulig.
Ved Norman-Roberts syndrom med lissensefali type I observeres kraniofasiale anomalier: alvorlig mikrocefali, lav pannehelling og utstående bred neserygg, vidtstående øyne (hyperterlorisme), underutvikling av kjevene (mikrognati). [ 23 ]
Miller-Dieker syndrom kan også kjennetegnes av en unormalt liten hodestørrelse med en bred, høy panne og kort nese, fordypninger i tinningene (bitemporale fordypninger) og lavtstående, deformerte ører.
Alvorlig lissencefalisyndrom er preget av mikrocefali, en reduksjon i størrelsen på øyeeplene (mikroftalmi), kombinert med retinal dysplasi, obstruktiv hydrocephalus og fraværende eller hypoplastisk corpus callosum.
Komplikasjoner og konsekvenser
Blant komplikasjonene ved denne anomalien nevner spesialister forstyrrelser i svelgefunksjonen (dysfagi) og gastroøsofageal refluks; ildfast (ukontrollert) epilepsi; hyppige øvre luftveisinfeksjoner; lungebetennelse (inkludert kronisk aspirasjon).
Spedbarn med lissensfali kan ha medfødte organiske hjerteproblemer i form av en atrieseptumdefekt eller en kompleks hjertefeil med cyanose (Fallots tetralogi). [ 24 ]
Konsekvensene av postnatal vekstretardasjon resulterer i de fleste tilfeller i død innen 24 måneder etter fødselen.
Diagnostikk lissencefali
Diagnosen starter med en fysisk undersøkelse av barnet, en studie av foreldrenes sykehistorie og graviditets- og fødselshistorie.
Under svangerskapet kan cellefri DNA-testing av fosteret, fostervannsprøve eller prøvetaking av korionvillus være nødvendig. [ 25 ] For mer informasjon, se – Prenatal diagnose av medfødte sykdommer
Instrumentell diagnostikk brukes til å visualisere hjernestrukturer og vurdere deres funksjoner:
- computertomografi av hjernen;
- magnetisk resonansavbildning (MR) av hjernen;
- elektroencefalogram (EEG). [ 26 ]
Under graviditet kan man mistenke lissensfali på ultralyd av fosteret etter 20–21 uker i fravær av parieto-occipitale og calcarine riller og en anomali i Sylvian-fissuren i hjernen.
Differensiell diagnose
Differensialdiagnostikk med andre syndromer av medfødte hjernedefekter utføres.
Det finnes mer enn 20 typer lissencefali, hvorav de fleste faller inn i to hovedkategorier: klassisk lissencefali (type 1) og brosteinslissencefali (type 2). Hver kategori har lignende kliniske manifestasjoner, men forskjellige genetiske mutasjoner.[ 27 ]
Hjerneundersøkelse ved type I lissencefali viser en hjernebark med fire lag i stedet for de seks man ser hos normale pasienter, mens ved type 2 lissencefali er hjernebarken uorganisert og fremstår klumpete eller nodulær på grunn av fullstendig forskyvning av hjernebarken av klynger av kortikale nevroner atskilt av gliomenkymalt vev. Pasientene hadde også muskel- og øyeavvik.
- Klassisk lissensfali (type 1):
- LIS1: Isolert lissencefali og Miller-Dieker syndrom (lissencefali assosiert med ansiktsdysmorfisme). [ 28 ]
- LISX1: DCX-genmutasjon. Sammenlignet med lissensfali forårsaket av LIS1-mutasjoner, viser DCX en sekslags cortex i stedet for fire.
- Isolert lissencefali uten andre kjente genetiske defekter
- Brosteinslissencefali (type 2):
- Walker-Warburg syndrom
- Fukuyama syndrom
- Sykdom i muskler, øyne og hjerne
- Andre typer kan ikke plasseres i en av de to gruppene ovenfor:
- LIS2: Norman-Roberts syndrom, lik lissencefali type I eller Miller-Dieker syndrom, men uten sletting av kromosom 17.
- LIS3
- LISX2
Mikrolissensfali: Dette er en kombinasjon av fravær av normal kortikal folding og et unormalt lite hode. Barn med regelmessig lissensfali har hoder av normal størrelse ved fødselen. Barn med en liten hodestørrelse ved fødselen diagnostiseres vanligvis med mikrolissensfali.
Det er også viktig å skille mellom lissencefali og polymikrogyri, som er forskjellige hjerneutviklingsdefekter.
Hvem skal kontakte?
Behandling lissencefali
Lissencefali er en uhelbredelig organisk defekt, så kun støttende og symptomatisk behandling er mulig. [ 29 ]
Først og fremst er dette bruk av antikonvulsiva og antiepileptika, samt installasjon av en gastrostomisonde i magen (hvis barnet ikke klarer å svelge selvstendig). Massasje er nyttig.
Ved alvorlig hydrocephalus fjernes cerebrospinalvæske.
Forebygging
Eksperter anbefaler at fremtidige foreldre søker genetisk veiledning, og at gravide registrerer seg hos fødselsleger og gynekologer i tide og gjennomgår alle planlagte undersøkelser.
Prognose
For barn med lissensfali avhenger prognosen av graden, men oftest overstiger ikke barnets mentale utvikling nivået på fire til fem måneder. Og alle barn med denne diagnosen lider av alvorlige psykomotoriske forstyrrelser og vanskelig behandlelig epilepsi. [ 30 ]
Ifølge NINDS (American National Institute of Neurological Disorders and Stroke) er den maksimale forventede levealderen for personer med lissencefali omtrent 10 år.