
Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
Rabies hos barn
Medisinsk ekspert av artikkelen

Sist anmeldt: 04.07.2025
Rabies, eller hydrofobi, er en akutt virussykdom som overføres gjennom bitt av et infisert dyr, med skade på nervesystemet og utvikling av alvorlig hjernebetennelse med dødelig utgang.
Epidemiologi
Rabiesviruset, som har vært en folkehelseplage siden antikken, forårsaker for tiden omtrent 59 000 dødsfall hvert år, hvorav nesten alle overføres via hundebitt. Dette har en betydelig økonomisk innvirkning på utviklingsland, spesielt i Afrika og Asia, som kan bære minst slike tap. Til tross for en dødelighet på nesten 100 % er hunderabies en fullstendig forebyggbar sykdom, og historiske eksempler på utryddelse av hunderabies i den utviklede verden vitner om dette. [ 1 ]
Fører til rabies
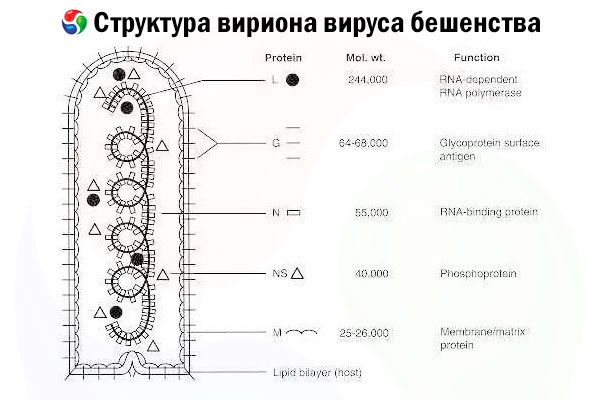
Den forårsakende agensen er rabiesviruset (RV), et negativt RNA-virus i rhabdovirusfamilien, omtrent 60 nm × 180 nm i størrelse.
Den består av en indre proteinkjerne, eller nukleokapsid, som inneholder nukleinsyre, og en ytre membran, et lipidholdig dobbeltlag dekket med transmembrane glykoproteinpigger. Den har en relativt enkel modulær genomstruktur og koder for fem strukturelle proteiner:
- RNA-avhengig RNA-polymerase (L),
- nukleoprotein (N),
- fosforylert protein (P),
- matriksprotein (M) og
- ytre overflate glykoprotein (G).
N-, P- og L-proteinene danner sammen med det genomiske RNA-et ribonukleoproteinkomplekset. G er det eneste RV-antigenet som er i stand til å indusere produksjonen av RV-nøytraliserende antistoffer, som er de viktigste immuneffektorene mot dødelig RV-infeksjon. På den annen side har ribonukleoproteinkomplekset vist seg å være det viktigste RV-antigenet som er i stand til å indusere CD4+ T-celler, som kan forbedre produksjonen av RV-nøytraliserende antistoffer gjennom intrastrukturell antigengjenkjenning.[ 2 ] Ribonukleoproteinkomplekset kan spille en viktig rolle i etableringen av immunologisk hukommelse og langsiktig immunitet.[ 3 ]
 [
[ Klassifisering og antigentyper
Slekten Lyssavirus omfatter rabiesvirus og antigenisk og genetisk beslektede rabiesvirus: Lagos-, Mokola- og Duvenhage-flaggermusvirus, samt to antatte undertyper av europeiske flaggermuslyssavirus. Kryssbeskyttelsesstudier indikerer at dyr som er immunisert med tradisjonelle rabiesvaksiner kanskje ikke er fullt beskyttet når de utfordres med andre lyssavirus.
Rabiesvirus kan klassifiseres som fikserte (tilpasset ved overføring i dyr eller cellekultur) eller gatevirus (villtype). Bruken av monoklonale antistoffer og genetisk sekvensering for å differensiere gatevirus har bidratt til å identifisere virusvarianter som stammer fra store vertsreservoarer over hele verden og til å antyde sannsynlige kilder til menneskelig eksponering når en historie med et definitivt dyrebitt ellers var fraværende hos en pasient.[ 8 ]
Patogenesen
Hovedreservoaret og smittekilden blant ville dyr er ulver, rever, sjakaler, flaggermus, og blant husdyr - hunder og katter, sjelden - hester, storfe, griser, rotter, osv. Overføring av smitte fra person til person, selv om mulig, er ekstremt sjelden. Dette er en typisk zoonotisk infeksjon. Mennesker blir smittet med rabies hovedsakelig fra hunder.
Etter at et menneske blir bitt av et sykt dyr, multipliserer viruset seg i muskelvevet på bittstedet, og når deretter endene av de sensoriske perifere nervene, sprer det seg sentripetalt og når motorneuronene. Tiden det tar for viruset å bevege seg og hjernen å bli påvirket, avhenger av bittstedet. Ved alvorlige bitt i hode og ansikt kan viruset nå sentralnervesystemet i løpet av 15–20 dager, og ved mindre skader på huden på overkroppen og lemmene, og dermed en liten dose av patogenet, kan prosessen med å flytte viruset til sentralnervesystemet bli forsinket i flere måneder eller til og med opptil 1–1,5 år. Etter å ha nådd sentralnervesystemet, fester viruset seg i vev i hjernen og ryggmargen, hovedsakelig i nevronene i medulla oblongata, ammoniumhornet og hjernebunnen. I ryggmargen er de bakre hornene mest påvirket. Fra sentralnervesystemet når viruset sentrifugalt langs nervestammene spyttkjertlene, hvor det multipliserer og skilles ut med spytt.
Konsepter i patogenesen til rabies
RV har et bredt vertsområde og kan infisere nesten alle pattedyr. Selv om det er rapportert om flere måter å overføre RV-viruset på, skjer naturlig smitte oftest via bitt. I tillegg til bitt kan inntak av RV-infiserte kadaver fremme rabiesvirusinfeksjon hos fjellrev, og kontakt mellom RV og slimhinner har vist seg å være en annen mulig smittevei.[ 9 ] Under noen uvanlige omstendigheter, som utilsiktet utslipp av RV som en aerosol i et laboratorium eller RV som en aerosol i huler bebodd av et stort antall flaggermus,[ 10 ] kan aerosoloverføring forekomme.
Det er ennå ikke klart om gate-RV og musetilpassede eller vevskulturtilpassede RV-stammer replikerer seg på inokuleringsstedet før de kommer inn i sentralnervesystemet. Mens eksperimentell intramuskulær infeksjon av unge hamstere eller vaskebjørner med gate-RV avslørte RV-replikasjon i tverrstripete muskelceller før viruset invaderte motoriske nevronaksoner over nevromuskulære overganger,[ 11 ],[ 12 ] viste intramuskulær infeksjon av mus med musetilpasset CVS-24 RV at RV migrerer direkte til sentralnervesystemet uten forutgående replikasjon på inokuleringsstedet.[ 13 ] Når RV er i terminalene til umyeliniserte aksoner, transporteres den retrograd til cellekroppen.
Nyere funn tyder på at aksonal vesikkeltransport kan representere en nøkkelstrategi for langdistanse virionbevegelse i aksoner.[ 14 ] Det har blitt anslått at RV migrerer innenfor aksoner med en hastighet på 3 mm/t.[ 15 ] Infeksjonen sprer seg deretter gjennom en kjede av nevroner forbundet med synaptiske forbindelser. Imidlertid er den nøyaktige mekanismen som fremmer transsynaptisk spredning fortsatt ukjent. Etter å ha infisert hjernen, sprer viruset seg sentrifugalt til det perifere og autonome nervesystemet i mange perifere organer.[ 16 ] I den siste fasen av infeksjonssyklusen migrerer RV til spyttkjertlene. Etter replikasjon i mukogene acinære celler frigjøres det i spytt og er klar for overføring til neste vert.[ 17 ]
Når det gjelder rabiesvirusindusert patologi, har apoptotisk celledød blitt foreslått som en potensiell patogen mekanisme i eksperimentelle rabiesmodeller av mus infisert med en fast stamme av RV.[ 18 ] En patogen mekanisme som kan bidra til den dype CNS-dysfunksjonen som er karakteristisk for rabies, kan være nedsatt nevronfunksjon. Genuttrykk har vist seg å være markant redusert i RV-infiserte nevroner, noe som resulterer i en generell undertrykkelse av proteinsyntese,[ 19 ] og flere studier har vist nedsatt nevrotransmisjon etter RV-infeksjon. Jiang demonstrerte at binding av en acetylkolinreseptorantagonist til infiserte rottehjernehomogenater var redusert sammenlignet med kontrollpersoner.[ 20 ] Nedsatt frigjøring og binding av serotonin, en nevrotransmitter involvert i kontrollen av søvnsyklus, smerteoppfatning og atferd, ble også observert i RV-infisert rottehjerne. [ 21 ], [ 22 ] I tillegg til å påvirke nevrotransmisjonen, kan høyre ventrikkelinfeksjon også påvirke ionekanaler. Infiserte musenevroblastomceller viser redusert funksjonell uttrykk av spenningsstyrte natriumkanaler, noe som kan forhindre aksjonspotensialer og til slutt føre til funksjonsnedsettelse. [ 23 ]
I tillegg til fraværet av alvorlige patologiske lesjoner i sentralnervesystemet (CNS), fremkaller de fleste tilfeller av rabies hos mennesker ikke en immunrespons 7 til 10 dager etter at kliniske tegn har oppstått. Disse betydelige forskjellene mellom patogenesen til rabies og de fleste andre virale eller bakterielle CNS-infeksjoner støttes ytterligere av det faktum at immunsuppresjon enten er ineffektiv eller skadelig for utfallet av rabies.[ 24 ] Det lave nivået av immunrespons som ofte observeres hos rabiesofre er forvirrende fordi det ikke kan forklares med den dårlige immunogenisiteten til RV-antigener. Faktisk er RV G og nukleokapsidprotein potente B- og T-celleantigener når de administreres parenteralt. [ 25 ] En mulig forklaring på den lave graden av immunrespons mot RV hos mennesker eller dyr med rabies kan være at RV-infeksjon i CNS forårsaker immunsuppresjon, [ 26 ] og det har blitt foreslått at RV bruker en subversiv strategi, inkludert å forhindre apoptose og ødelegge invaderende T-celler. [ 27 ]
Svekkede RV-stammer som har blitt tilpasset ikke-nevronale celler, skiller seg betydelig fra patogene gate-RV-stammer i sin nevroinvasivitet, som refererer til deres evne til å invadere CNS fra perifere steder. I denne forbindelse mangler eller har vevskulturtilpassede RV-stammer bare begrenset evne til å invadere CNS fra perifere steder, mens gate-RV-stammer eller mustilpassede RV-stammer som CVS-24 er svært invasive.[ 28 ] Viktige faktorer involvert i RV-nevroinvasjon inkluderer virusopptak, aksonal transport, transsynaptisk spredning og virusreplikasjonsrate.
Inntil nylig var vår kunnskap om RV-patogenese begrenset og var hovedsakelig basert på beskrivende studier av RV-stammer på gaten eller eksperimentelle infeksjoner med svekkede stammer tilpasset i laboratoriet. Fremveksten av omvendt genetikkteknologi har gjort det mulig for oss å identifisere de virale elementene som bestemmer den patogene fenotypen til RV og å bedre forstå mekanismene involvert i rabiespatogenese.
Identifisering av virale elementer som kontrollerer opptak, spredning og replikasjon av rabiesvirus
- Virale elementer involvert i virusfangst
RV-infeksjon begynner med at viruset fester seg til en antatt cellulær reseptor. Selv om flere membranoverflatemolekyler har blitt foreslått som RV-reseptorer, inkludert nikotinacetylkolinreseptoren,[ 29 ] det nevrale celleadhesjonsmolekylet[ 30 ] og lavaffinitetsnevrotrofinreseptoren p75 NTR,[ 31 ] er det fortsatt uklart om disse molekylene faktisk spiller en rolle i rabiesvirusets livssyklus. I denne sammenhengen har det nylig blitt vist at RV G–p75 NTR-interaksjonen ikke er nødvendig for RV-infeksjon av primære nevroner.[ 32 ] Etter reseptorbinding internaliseres RV via adsorptiv eller reseptormediert endocytose. [ 33 ] Det lave pH-miljøet i det endosomale kammeret induserer deretter konformasjonsendringer i RV G som utløser fusjon av virusmembranen med den endosomale membranen, og dermed frigjør RNP i cytoplasmaet. [ 34 ] For virus spiller RV G en kritisk rolle i virusopptak, mest sannsynlig gjennom interaksjoner med antatte cellulære reseptorer som muliggjør raskt opptak. I denne forbindelse har det blitt vist at patogeniteten til vevskulturtilpassede RV-stammer (f.eks. ERA, HEP og CVS-11) korrelerer med tilstedeværelsen av en determinant lokalisert i antigensted III av G-proteinet. [ 35 ] En Arg → Gln-mutasjon i posisjon 333 i dette antigenstedet av ERA G-proteinet resulterte i en syv ganger så lang forsinkelse i internaliseringen av Gln333 RV-varianten sammenlignet med villtypevarianten. Asn194→Lys194-mutasjonen i RV G, som forklarer gjenoppkomsten av den patogene fenotypen, var assosiert med en betydelig reduksjon i internaliseringstid.[ 36 ] Videre viste eksperimenter med kimære RV-er at tiden som kreves for internalisering av RV-virioner var betydelig økt og patogeniteten var sterkt redusert etter erstatning av G-genet til den høypatogene SB RV-stammen, som var avledet fra en cDNA-klon av den sølvavledede flaggermus-assosierte stammen RV-18,[ 37 ] med den høyt svekkede SN-stammen, som ble isolert fra en cDNA-klon av SAD B19 RV-vaksinestammen.[ 38 ] Sammen støtter disse dataene antagelsen om at kinetikken til virusopptak, som er en funksjon av RV G, er en viktig determinant for RV-patogenitet.
- Virale elementer involvert i spredning og overføring av virus
En unik egenskap ved rabiesvirus er dets evne til å spre seg fra celle til celle. Observasjonen om at Gln333 ERA-varianten mister pH-avhengig celle-celle-fusjonsaktivitet in vitro [ 39 ] og viser en sterkt redusert evne til å spre seg fra celle til celle [ 40 ], antyder at RV G også spiller en nøkkelrolle i celle-til-celle-spredning og dermed virusoverføring, sannsynligvis gjennom sin fusiogene aktivitet. Denne muligheten støttes ytterligere av funnet om at spredningsraten til den patogene RV-revertanten SPBNGAK er nesten dobbelt så høy som den som er bestemt for den ikke-patogene SPBNGA-varianten. Interessant nok forårsaket Asn 194 → Lys 194-mutasjonen i G SPBNGAK en forskyvning i pH-terskelen for membranfusjon til en høyere pH, noe som støtter hypotesen om at en høyere pH-terskel for membranfusjon er assosiert med økt virusspredning. [ 41 ]
Studier av transnevronale indikatorer på RV-infeksjon hos rotter [ 42 ] og rhesusaper [ 43 ] har vist at rabiesvirus migrerer utelukkende i retrograd retning i aksoner. Selv om flere RV-proteiner er involvert i nevrale transportmekanismer, ser det ut til at RV G spiller en dominerende rolle i transnevronal spredning av RV-infeksjon. For eksempel, mens perifer infeksjon med equine infectious anemia virus (EIAV) pseudotypet med RV G resulterer i virusoverføring til ryggmargen, klarte ikke den samme EIAV pseudotypet med vesikulær stomatittvirus G å komme inn i nervesystemet. [ 44 ] Videre ble virusspredningen av ERA G Arg 333 → Gln 333-mutanten i CNS funnet å være sterkt redusert sammenlignet med villtypemutanten, noe som ytterligere tyder på en funksjon av intakt RV G i transsynaptisk spredning. Det mest overbevisende beviset for en viktig rolle for RV G i transsynaptisk transport kommer imidlertid fra intrakraniell infeksjon av mus med et rekombinant G-defekt RV-virus, som viste at infeksjonen forble begrenset til nevroner på inokuleringsstedet uten tegn til spredning til sekundære nevroner.[ 45 ] Det er imidlertid sannsynlig at i tillegg til RV G, spiller RV M også en rolle i virusspredning og dermed i transsynaptisk transport. I denne forbindelse ble det vist at spredningen av den kimære SN-BMBG RV-varianten, som inneholder både M og G fra den høypatogene SB, var betydelig høyere enn spredningen av den kimære SN-BG- eller SN-BM-varianten, som inneholder henholdsvis G og M fra SB, noe som tyder på at optimal interaksjon mellom M og G kan spille en viktig rolle i celle-til-celle-virusspredning. [ 46 ] Siden RV M støtter virusknoppskyting, [ 47 ] er det sannsynlig at den mer effektive spredningen av den kimære RV SN-BMBG-varianten skyldes optimal virusknoppskyting ved den postsynaptiske membranen.
Nyere studier har vist at samspillet mellom RV P og den lette dyneinkjeden knytter RV RNP til vertscellens transportsystem, og dermed letter retrograd aksonal transport av viruset.[ 48 ],[ 49 ] Perifer infeksjon av voksne mus viste imidlertid at sletting av LC8-bindingsdomenet til RV P ikke forhindrer virusinntreden i sentralnervesystemet, noe som tyder på at RV-proteinet ikke er direkte involvert i retrograd aksonal spredning av RV.[ 50 ]
- Virale elementer som kontrollerer viral replikasjon
I motsetning til mange andre virus, som influensavirus, er RV-patogenisiteten omvendt proporsjonal med hastigheten på viral RNA-syntese og produksjon av smittsomme viruspartikler. Sammenligning av viralt mRNA og genomisk RNA-nivåer produsert av forskjellige kimære virus antyder at viral RNA-transkripsjon og replikasjon reguleres av flere faktorer, inkludert RV M, som har blitt identifisert som en transaktiv faktor som medierer overgangen fra initialt høye nivåer av mRNA-syntese til genomisk RNA-syntese.[ 51 ] Videre er M fra alle rhabdovirus i stand til å stenge av viral genuttrykk ved å binde seg til RNP, noe som resulterer i dannelsen av en svært kondensert ryggradlignende struktur som ikke er i stand til å støtte RNA-syntese.
For å identifisere andre virale elementer som kontrollerer patogenitet ved å regulere viral replikasjon, ble de 5'-terminale sekvensene til den høypatogene SB-stammen erstattet trinnvis med sekvenser fra den høyt svekkede SN-vaksinestammen, noe som resulterte i rekombinante virus SB2 (terminal sekvens [TS] + L), SB3 (TS + L + pseudogen [Ψ]), SB4 (TS + L + Ψ + G) og SB5 (TS + L + Ψ + G + M). Intramuskulær infeksjon med foreldrevirusene SB og SN og de kimære RV-ene SB2, SB3, SB4 og SB5 forårsaket de høyeste dødelighetsratene hos SB-infiserte mus og ingen sykelighet eller dødelighet hos SN-infiserte mus. Erstatning av TS, L og SB med de tilsvarende elementene fra SN resulterte i en beskjeden reduksjon i sykelighet og dødelighet, og en ytterligere G eller G pluss M-utveksling reduserte kraftig eller fullstendig viruspatogenitet.
Fenotypisk karakterisering av disse villtype- og kimære RV-ene i vevskultur viste at patogeniteten til en gitt RV er omvendt korrelert med dens evne til å replikere i nevronceller. Selv om SB replikerte på nivåer nesten 1000 ganger lavere enn SN, og erstatning av TS, L og i SB med SN-nivåer hadde liten effekt på virusvekstkinetikken, resulterte ytterligere erstatning av G eller G pluss M av SB med de tilsvarende SN-genene i en 1-log økning i virusproduksjon, noe som tyder på at viralt RNA-replikasjonskinetikk så vel som viruspartikkelproduksjon i stor grad kontrolleres av RV G-proteinet. Denne konklusjonen støttes av data innhentet med RV G-varianter som avviker med én aminosyre i G-proteinene deres. Den patogene rabiesvirusvarianten SPBNGAK 194 produserte en virustiter i NA-celler som var 1 log lavere enn den som ble produsert av den ikke-patogene varianten SPBNGAK 194, og sanntids-PCR-analyse viste at ratene for viral RNA-transkripsjon og -replikasjon i SPBNGAK-infiserte NA-celler var 5 og 10 ganger høyere enn i SPBNGAK-infiserte NA-celler.[ 52 ] Ytterligere bevis for en invers korrelasjon mellom patogenitet og ratet for viral RNA-syntese og viral partikkelproduksjon ble gitt av mus infisert med kimære rekombinante virus der G- og M-genene til den svekkede SN-stammen ble erstattet av de fra den høypatogene SB-stammen. Disse eksperimentene viste en betydelig økning i patogeniteten til den foreldre SN-stammen som bar RV G i forhold til den patogene SB-stammen. Patogeniteten ble ytterligere økt når både G og M fra SB ble introdusert i SN.
Substitusjon av G eller M eller begge i SN med de tilsvarende genene fra SB var assosiert med en signifikant reduksjon i hastigheten på viruspartikkelproduksjon samt hastigheten på viral RNA-syntese. Disse dataene indikerer at både G og M spiller viktige roller i RV-patogenesen ved å regulere virusreplikasjon. Funnet om at substitusjon av G eller G pluss M i SN med G eller G pluss M av SB resulterer i en moderat til sterk reduksjon i henholdsvis viral RNA-transkripsjon og -replikasjon, mens substitusjon av M alene i SN med M av SB resulterer i en sterk økning i viral RNA-transkripsjon og -replikasjon, indikerer at RV G også har en viktig regulatorisk funksjon i viral RNA-transkripsjon/replikasjon enten alene eller gjennom interaksjon med M-proteinet. Mekanismen som RV G-genet kontrollerer viral RNA-syntese med er ukjent. Visse nukleotidsekvenser i RV G-genene, slik som de som inkluderer kodonene for Arg333 og Lys 194, har blitt identifisert som mål for cellulære miRNAer. Det har blitt vist at målgjenkjenning av cellulære miRNAer kan resultere i positiv eller negativ regulering av virusreplikasjon. [ 53 ] Arg 333 → Glu 333 eller Lys 194 → Ser 194-substitusjoner i RV G-gensekvensen resulterer i avskaffelse av miRNA-målsekvenser, som igjen er assosiert med en betydelig økning i hastigheten på viral RNA-syntese [Faber M, Thomas Jefferson University, PA, USA, upubliserte data], noe som tyder på at vertscellulære miRNA-er også spiller en viktig rolle i reguleringen av RV-replikasjon, slik det har blitt vist for andre RNA-virus, inkludert vesikulært stomatittvirus og HCV. [ 54 ], [ 55 ]
Regulering av virusreplikasjon ser ut til å være en av de viktigste mekanismene involvert i RV-patogenesen. For å unngå immunresponsen og bevare integriteten til det nevrale nettverket, kan patogene RV-stammer, men ikke svekkede stammer, regulere vekstraten deres. En lavere replikasjonsrate er sannsynligvis til fordel for patogene RV-stammer ved å bevare den nevrale strukturen som disse virusene bruker for å nå sentralnervesystemet. En annen forklaring på den lavere replikasjonsraten for patogen RV er at viruset, for å unngå tidlig deteksjon av vertens immunsystem, opprettholder minimale nivåer av uttrykk av antigenene sine.
Forholdet mellom RV G-ekspresjon, apoptose og patogenitet
Det er velkjent at street rabiesvirusstammer som er betydelig mer patogene enn vevskulturtilpassede stammer uttrykker svært begrensede nivåer av G og ikke induserer apoptose før sent i den infeksjonelle syklusen, noe som tyder på at patogeniteten til en bestemt virusstamme er omvendt korrelert med RV G-ekspresjon og evnen til å indusere apoptose.[ 56 ] Direkte bevis for en korrelasjon mellom nivået av G-ekspresjon og omfanget av apoptose ble oppnådd med den rekombinante RV SPBNGA-GA, som bar to identiske G-gener og overuttrykte RV G.[ 57 ] Morfologiske studier av nevronkulturer infisert med denne rekombinante RV viste at celledød var betydelig økt parallelt med RV G-overekspresjon, og at apoptose er den viktigste mekanismen involvert i RV G-mediert død. Spesielt er reduksjonen i F-aktinfarging etter SPBNGA-GA-infeksjon i samsvar med apoptoseindusert depolymerisering av aktinfilamenter. Videre var antallet TUNEL-positive kjerner i SPBNGA-GA-infiserte nevroner betydelig økt sammenlignet med det i uinfiserte og SPBNGA-infiserte nevroner. Mekanismen som RV G-genet medierer den apoptotiske signaleringsprosessen gjennom, er imidlertid fortsatt stort sett ukjent. Det har blitt antydet at RV G-ekspresjon over en viss terskel alvorlig forstyrrer cellemembranen. Det er høyst sannsynlig at apoptotiske celler ikke elimineres raskt i CNS og derfor gjennomgår sekundær nekrose. [ 58 ] På den annen side kan RV-infeksjon, og spesielt overekspresjon av RV G-protein, føre til pyroptose, en celledødsvei som ligner på apoptose, som i motsetning til apoptose involverer aktivering av caspase 1 og dermed fører til nekrose. [ 59 ] Graden av nekrose eller pyroptose indusert av RV-infeksjon spiller sannsynligvis en kritisk rolle i induksjonen av antiviral immunitet. Mens apoptotiske celler opprettholder sin membranintegritet og ikke stimulerer den medfødte immunresponsen, blir nekrotiske celler permeabiliserte og utskiller endogene adjuvanser som kan utløse en robust medfødt immunrespons. [ 60 ]
Siden nivået av apoptose/nekrose korrelerer med RV-immunogenisitet, har det blitt foreslått at den immunstimulerende effekten av apoptotiske/nekrotiske celler mest sannsynlig bidrar til generering av en beskyttende immunrespons. Derfor er regulering av RV G-ekspresjon sannsynligvis en viktig faktor i rabiespatogenesen, ettersom det gir et middel for overlevelse og spredning av patogene RV-varianter i nervesystemet uten å forårsake åpenbar nevronskade og fremkalle en beskyttende immunrespons som ville forhindre infeksjon.
RV G-ekspresjon kan reguleres på nivået av RNA-syntese, post-translasjonelt nivå, eller begge deler. Nivåene av RV G uttrykt av forskjellige kimære RV-varianter har vist seg å være reflektert av hastigheten på viral RNA-syntese, noe som tyder på at ulik regulering av RV G-ekspresjon av disse variantene skyldes variasjoner i hastigheten på viral mRNA-transkripsjon. Som med viral RNA-transkripsjonshastigheter, korrelerer mengden RV G uttrykt av disse variantene omvendt med viral patogenitet. På den annen side resulterte infeksjon av primære nevronkulturer med den mindre patogene RV-varianten CVS-B2c i fire ganger høyere nivåer av G-protein enn infeksjon med den høypatogene varianten CVS-N2c, til tross for syntesen av sammenlignbare nivåer av G mRNA i begge infeksjonene. Puls-chase-eksperimenter viste at de høyere G-proteinnivåene i CVS-B2c-infiserte nevroner i stor grad var et resultat av en lavere nedbrytningshastighet av CVS-B2c G-proteinet sammenlignet med CVS-N2c G-proteinet. Mekanismen som fører til den raskere proteolytiske nedbrytningen av CVS-N2c G-proteinet gjenstår imidlertid å belyse.
Symptomer rabies
Inkubasjonstiden for rabies er i gjennomsnitt 30–90 dager. Ved massiv infeksjon gjennom store sår i hode og ansikt kan den forkortes til 12 dager. I sjeldne tilfeller kan inkubasjonstiden vare i 1 år eller mer.
Det er en strengt sekvensiell endring av tre perioder av sykdommen: prodromal, eksitasjon, lammelse.
Prodromperioden begynner med verkende eller trekkende smerter på bittstedet, samt smerter langs nervene. I arrområdet kan det være en brennende følelse, kløe, noen ganger rødhet og hevelse. Pasienten opplever generell uvelhet, hodepine, kvalme. Oppkast, økning i kroppstemperatur til 37,5-38 °C og symptomer på en progressiv psykisk lidelse er observert: økt reflekseksitabilitet, en uforklarlig følelse av angst, frykt, melankoli. Ofte er pasienten deprimert, hemmet, tilbaketrukket, nekter å spise, sover dårlig, klager over dystre tanker, skremmende drømmer. Prodromperioden varer 2-3 dager, noen ganger strekker den seg til 7 dager. På slutten av denne perioden kan det være anfall av angst med kortvarige pustevansker, en følelse av tetthet i brystet, ledsaget av takykardi og økt respirasjonsfrekvens.
Perioden med opphisselse er preget av forekomsten av hydrofobi: når pasienten prøver å drikke, og deretter ved synet av vann eller en påminnelse om det, opplever vedkommende en krampaktig spasme i svelget og strupehodet, der vedkommende kaster vannkoppen med et skrik, kaster skjelvende hender fremover, kaster hodet og kroppen bakover. Nakken er strukket ut, en smertefull grimase forvrenger ansiktet, som blir blålig på grunn av spasmer i pustemusklene. Øynene buler ut, uttrykker frykt, ber om hjelp, pupillene utvides, pupillene er vanskelige å puste inn. På høyden av anfallet er hjerte- og respirasjonsstans mulig. Anfallet varer i flere sekunder, hvoretter pasientens tilstand ser ut til å bli bedre. Deretter kan anfall av spasmer i musklene i strupehodet og strupehodet oppstå selv fra luftbevegelse (aerofobi), sterkt lys (fotofobi) eller et høyt ord (akustikofobi). Anfallene er ledsaget av psykomotorisk agitasjon, der pasienten oppfører seg som en "galning". Bevisstheten er uklar under anfallet, men klarner opp i den interiktale perioden. I løpet av agitasjonsperioden, på grunn av økt tonus i det sympatiske nervesystemet, opplever pasientene en kraftig økning i spyttsekresjon (sialoré) med manglende evne til å svelge spytt på grunn av spasmer i svelgmusklene. Pasienten spruter spytt. Noen pasienter kan utvikle tegn på meningisme og til og med opistotonus, og kramper er vanlige. I dette tilfellet kan cerebrospinalvæsken ikke endre seg, men hos noen pasienter kan proteinkonsentrasjonen øke og antallet celler kan øke på grunn av lymfocytter.
Uten tilstrekkelig behandling øker tegn på dehydrering, ansiktstrekkene blir skarpere og kroppsvekten synker. Kroppstemperaturen stiger til høye verdier. Krampetrekninger er mulige. Varigheten av eksitasjonsstadiet er omtrent 2-3 dager, sjelden 4-5 dager. Et dødelig utfall oppstår vanligvis under et av anfallene. I sjeldne tilfeller overlever pasienten til det tredje stadiet av sykdommen.
I løpet av lammelsesperioden roer pasienten seg ned. Anfallene av hydrofobi opphører, pasienten kan drikke og svelge mat, bevisstheten er klar. Til tross for det tilsynelatende velværet øker imidlertid sløvhet, apati og depresjon, lammelse av lemmene, bekkenforstyrrelser og lammelse av kranialnervene oppstår snart. Kroppstemperaturen stiger til 42–43 °C, arterietrykket synker, og innen utgangen av den første dagen inntreffer døden som følge av lammelse av kardiovaskulære og respiratoriske sentre.
Nøytrofil leukocytose, økt hemoglobin, erytrocytter og hematokrit observeres i perifert blod.
Hva plager deg?
Skjemaer
Klinisk skilles det mellom typiske og atypiske former. Atypiske former omfatter alle tilfeller uten opphisselse og hydrofobi. Atypiske former omfatter bulbær, cerebellær, meningoencefalittisk, etc.
Diagnostikk rabies
Påvisning av rabiesantigen, antistoffer, viralt RNA eller virusisolering muliggjør diagnosen rabies. Fordi enhver individuell test kan være negativ hos en pasient med rabies, er serielle serumprøver for påvisning av rabiesantistoffer, spyttprøver for viruskultur og hudbiopsi for direkte immunofluorescenstesting for viralt antigen noen ganger nødvendig, spesielt når rabiesmistanken er sterkt antatt.
En av de raskeste metodene for å diagnostisere rabies antemortem hos mennesker er å utføre en direkte immunofluorescenstest på en hudbiopsi i nakken for å oppdage rabiesantigen. Den direkte immunofluorescenstesten er den mest sensitive og spesifikke metoden for å oppdage rabiesantigen i hud og annet ferskt vev (f.eks. hjernebiopsi), selv om resultatene av og til kan være negative tidlig i sykdommen. Hvis ferskt vev ikke er tilgjengelig, kan enzymatisk fordøyelse av fiksert vev øke reaktiviteten til immunofluorescenstesten. Imidlertid kan sensitiviteten være uakseptabelt lav.
Diagnosen kan også stilles hvis viruset isoleres fra spytt etter inokulering av nevroblastomceller eller laboratoriegnagere; dette er vanligvis mest effektivt i løpet av de første 2–3 ukene av sykdommen. Påvisning av rabiesvirus-nøytraliserende antistoffer, vanligvis utført ved rask fluorescerende fokusinhibisjonstest (RFFIT), i serum fra uvaksinerte individer er også diagnostisk. Tilstedeværelsen av antistoffer i cerebrospinalvæsken bekrefter diagnosen, men de kan oppstå 2–3 dager senere enn serumantistoffer og kan derfor være mindre nyttige i de tidlige stadiene av sykdommen. Selv om den serologiske responsen etter vaksinasjon vanligvis ikke kan skilles fra den serologiske responsen indusert av sykdommen, produserer vaksinasjon vanligvis ikke antistoffer mot cerebrospinalvæsken.
Bare syv tilfeller av rabies-"tilfriskning" i løpet av de siste 25 årene er godt dokumentert. Selv om rabiesvirus ikke ble isolert fra noen av pasientene, støttet høye titere av rabiesnøytraliserende antistoffer i serumprøver og tilstedeværelsen av nøytraliserende antistoffer i cerebrospinalvæsken diagnosen sterkt.
Hva trenger å undersøke?
Hvilke tester er nødvendig?
Differensiell diagnose
Diagnosen rabies hos mennesker stilles vanligvis på grunnlag av epidemiologiske og kliniske data og bekreftes i laboratoriet. Diagnosen er enkel hvis det foreligger en historie med dyrebitt og hele spekteret av symptomer og tegn har forekommet. Ellers er en nøye, men rask evaluering av de epidemiologiske og kliniske trekkene ved mindre typiske tilfeller nødvendig før spesifikke laboratorietester utføres. Enhver pasient med nevrologiske tegn eller symptomer eller uforklarlig encefalitt bør avhøres om muligheten for eksponering for dyr i rabiesendemiske områder innenfor eller utenfor bostedslandet. Manglende mistanke om rabies i flere nylige menneskelige dødsfall i USA kan ha skyldtes mangel på nøye eksponeringshistorie.
Ved sykdomsutbruddet kan rabies ligne på mange smittsomme og ikke-smittsomme sykdommer. Mange andre encefalitter, som de som er forårsaket av herpesvirus og arbovirus, ligner på rabies. Andre smittsomme sykdommer kan også ligne på rabies, som stivkrampe, cerebral malaria, rickettsiose og tyfoidfeber. Paralytiske smittsomme sykdommer som kan forveksles med rabies inkluderer polio, botulisme og herpes simian B-encefalitt.
Ikke-smittsomme sykdommer som kan forveksles med rabies inkluderer en rekke nevrologiske syndromer, spesielt akutt inflammatorisk polynevropati (Guillain-Barré syndrom), samt allergisk encefalomyelitt etter vaksinasjon sekundært til rabiesvaksinasjon av nervevevet, forgiftning eller rusmiddelforgiftning, alkoholabstinenser, akutt porfyri og rabieshysteri. Guillain-Barré syndrom kan forveksles med paralytisk rabies, og omvendt.
Hvem skal kontakte?
Behandling rabies
Behandling for rabies er ikke utviklet. Administrering av store doser spesifikt anti-rabies immunoglobulin og leukocyttinterferon er ineffektivt. Symptomatisk behandling administreres for å lindre pasientens lidelse. For dette formålet plasseres pasienten på en egen avdeling eller boks, og det opprettes et beskyttende regime som begrenser påvirkningen fra det ytre miljøet (redusert støy, sterkt lys, luftstrøm). For å redusere sentralnervesystemets eksitabilitet foreskrives sovepiller, antikonvulsiva og smertestillende midler. Vannbalansen normaliseres.
I lammelsesstadiet foreskrives legemidler som stimulerer aktiviteten til kardiovaskulære og respiratoriske systemer. Det anbefales å bruke hyperbarisk oksygenering, cerebral hypotermi, kontrollert mekanisk pusting med fullstendig kurarisering av pasienten. Imidlertid er alle behandlingsmetoder praktisk talt ineffektive. I beste fall er det mulig å forlenge pasientens liv i flere måneder. Et ugunstig utfall er forhåndsbestemt av alvorlighetsgraden av skaden på hjernestammen med ødeleggelse av vitale sentre.
Forebygging
Utviklingen av den første rabiesvaksinen av Pasteur i 1885 innledet en æra med mye mer effektiv rabieskontroll. I dag, til tross for nesten 100 % dødelighet hos mennesker fra rabies, kan sykdommen forebygges fullstendig gjennom vaksinasjon før og/eller etter eksponering. Mens Pasteur og hans kolleger startet vaksinasjonen av private hunder i Paris, ble den første massevaksinasjonen av hunder utført tidlig på 1920-tallet i Japan, noe som markerte det første store nasjonale rabieskontrollprogrammet. Oral vaksinasjon av ville dyr, først utviklet på 1970-tallet, har siden gjentatte ganger vist seg å effektivt kontrollere sykdommen hos store landverter som rever, vaskebjørn og stinkdyr.[ 68 ] Vedvarende rabiesvaksinasjon av reservoardyrpopulasjoner med 70 % eller høyere dekningsgrad vil til slutt eliminere RABV fra reservoararter og forhindre spredning av viruset til tilfeldige verter. [ 69 ]
Fylogenetiske data indikerer at lyssavirus infiserte flaggermus lenge før de infiserte landpattedyr, og de fleste lyssavirus, inkludert RABV, sirkulerer fortsatt i ulike flaggermusarter over hele verden.[ 70 ] Effektive metoder for å forhindre overføring av RABV blant flaggermus er imidlertid fortsatt vanskelige å finne, noe som utelukker muligheten for fullstendig utryddelse av rabies på dette tidspunktet. Selv etter eksponering for RABV gjennom bitt av et rabiesinfisert pattedyr, kan imidlertid sikker og effektiv posteksponeringsprofylakse (PEP, inkludert sårrens, rabiesimmunglobulin og rabiesvaksinasjon) beskytte mennesker mot rabiesinfeksjon hvis behandlingen administreres raskt og i henhold til anbefalingene fra Verdens helseorganisasjon (WHO).
Disse to metodene for å forhindre menneskelige dødsfall – den ene basert på å vaksinere eksponerte personer og den andre basert på å vaksinere nok hunder til å bryte smittespiralen ved kilden – er byggesteinene i en «én helse»-tilnærming til forebygging og kontroll av rabies hos hunder. Disse to forskjellige måtene for å forhindre menneskelige dødsfall ble vurdert som separate alternativer: Strategi A, basert på å gi mennesker PEP, og strategi B, basert på å vaksinere hunder; eller som komponenter i en kombinert strategi A + B i en analyse av de sannsynlige kostnadene ved de alternative strategiene.[ 71 ]
Land som Thailand har hatt enorm suksess med å forhindre menneskelige dødsfall gjennom bruk av PEP, men har også opplevd økende etterspørsel og tilhørende kostnader knyttet til bruk av PEP alene. [ 72 ] For eksempel, sammenlignet med situasjonen i 1991, trengte fire ganger så mange mennesker (mer enn 400 000) PEP i 2003. Nyere data viser at Folkerepublikken Kina, som vaksinerer 15 millioner mennesker per år etter potensiell rabieseksponering, bruker omtrent 650 millioner amerikanske dollar per år på PEP alene. [ 73 ]
En mye mer bærekraftig tilnærming er å forhindre spredning av smitte ved kilden, i dyrepopulasjonen, samtidig som man øker tilgangen til PEP for eksponerte menneskelige pasienter når det er nødvendig. Der det er politisk vilje og tilstrekkelig finansiering til å kontrollere hunderabies, kan og har dødsfall blitt eliminert. Utbredt bruk av hundevaksinasjon har ført til utryddelse av hunderabies fra flere land, inkludert Malaysia i 1954, [ 74 ] Japan i 1956, Taiwan i 1961, Singapore, og spesielt i hele Vest-Europa (gjennomgått i Rupprecht et al., King et al., og Gongal og Wright). [ 75 ]