
Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
Aper-syndromet
Medisinsk ekspert av artikkelen

Sist anmeldt: 04.07.2025

 [
[ Fører til Aper-syndromet
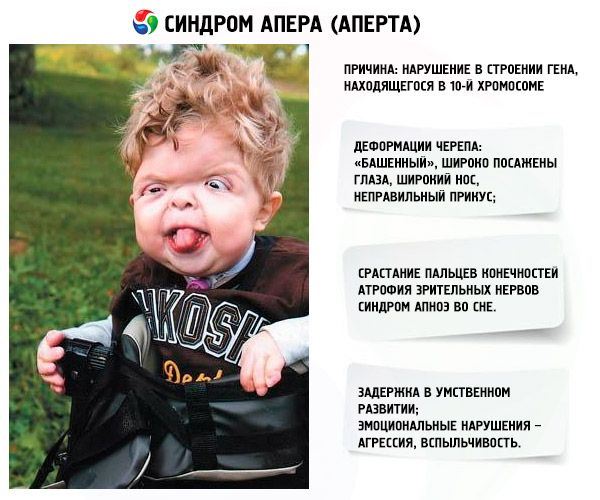
Utviklingen av syndromet provoseres av en forstyrrelse i strukturen til et gen som ligger på kromosom 10. Dette genet er ansvarlig for prosessen med fingerseparasjon, og i tillegg for rettidig lukking av kraniale suturer.
I tillegg anses smittsomme sykdommer som oppstår under graviditet (som røde hunder eller tuberkulose, syfilis eller influensa, samt hjernehinnebetennelse), og morens eksponering for røntgenstråler å være årsakene til patologien. Oftest oppdages et slikt syndrom hos barn født av eldre foreldre.
Patogenesen
Apert syndrom er arvelig. Typen er autosomal dominant (dvs. i en familie der en av de fremtidige foreldrene er syk, er sannsynligheten for å få et barn med denne sykdommen 50–100 %).
En unik mutasjon i fibroblastvekstfaktorreseptor 2 (FGFR2) resulterer i et økt antall progenitorceller som utvikler seg langs den osteogene banen. Dette resulterer til slutt i økt dannelse av subperiosteal benmatrise og for tidlig ossifikasjon av kraniale suturer under fosterutviklingen. Rekkefølgen og hastigheten på sutursmeltingen bestemmer graden av deformitet og funksjonshemming. Når suturmaterialet har grodd, er veksten av annet vev vinkelrett på denne suturen begrenset, og de sammenvokste beinene fungerer som et enkelt beinstillas.
Det første genetiske beviset på at syndaktyli ved Apert syndrom skyldes en defekt i keratinocyttvekstfaktorreseptoren (KGFR) var observasjonen av en korrelasjon mellom KGFR-ekspresjon i fibroblaster og alvorlighetsgraden av syndaktyli.
Amblyopi og strabismus er vanligere hos pasienter med FGFR2 Ser252Trp-mutasjonen, og diskatrofi på synsnerven er vanligere hos pasienter med FGFR2 Pro253Arg-mutasjonen. Pasienter med FGR2 Ser252Trp-mutasjoner har en betydelig høyere forekomst av synshemming sammenlignet med pasienter med FGFR2 Pro253Arg-mutasjonen.
Symptomer Aper-syndromet
Noen manifestasjoner av sykdommen er tydelig merkbare allerede ved fødselen av barnet, fordi de utvikler seg i mors livmor. Blant de viktigste symptomene på syndromet:
- Hodeskallen er deformert – den strekker seg i høyden og får et «tårnlignende» utseende; i tillegg er øynene bredt plassert og litt utstående (fordi størrelsen på øyehulene avtar); nesen utvides og det dannes et feil bitt (de øvre tennene stikker for mye ut);
- Fingrene på lemmene er fullstendig sammenvokst (hovedsakelig ringfingeren, samt langfingeren og pekefingeren) og ser ut som en hudmembran eller en fullstendig fusjon av bein; i tillegg kan ekstra fingre vokse;
- Psykisk utviklingshemming (forekommer ikke hos alle);
- Synsnervene atrofierer, noe som resulterer i redusert synsskarphet (i noen tilfeller utvikler fullstendig synstap);
- Økt intrakranielt trykk, som oppstår som følge av for tidlig lukking av kraniale suturer, manifesterer seg i form av hodepine og oppkast med kvalme;
- Siden overkjeven forblir underutviklet, oppstår pusteproblemer;
- Søvnapnésyndrom er en vanlig forekomst.
- Emosjonelle manifestasjoner – aggresjon, mangel på selvkontroll, sterk irritabilitet.
Komplikasjoner og konsekvenser
De fleste pasienter har en viss grad av obstruksjon av de øvre luftveiene i spedbarnsalderen. De øvre luftveiene er dårlig patenterte på grunn av den reduserte størrelsen på nesesvelget og nesehinnene, og de nedre luftveiene kan forårsake tidlig død på grunn av abnormaliteter i luftrøret.
Diagnostikk Aper-syndromet
For å stille en diagnose kreves følgende trinn:
- Legen bør analysere pasientens klager, samt sykehistorien. Det er nødvendig å finne ut om det har vært tilfeller av lignende patologi i familien;
- Nevrologisk undersøkelse for å vurdere hodeskallens form og pasientens intellektuelle utvikling (spesielle spørreskjemaer samt intervju);
- Undersøkelse av fundus for å oppdage symptomer på økt intrakranielt trykk (hevelse i optisk disk, samt uskarphet i kantene);
- For å vurdere tilstanden til hodeskallen utføres et røntgenbilde;
- Datamaskin- og magnetisk resonansavbildning av hodet for å undersøke hjernens og hodeskallens struktur lag for lag, for å bestemme tilstedeværelsen av symptomer på for tidlig fusjon av kraniale suturer, og i tillegg hydrocephalus (på grunn av økt intrakranielt trykk akkumuleres overflødig cerebrospinalvæske (dette er cerebrospinalvæsken som fremmer metabolske prosesser, samt ernæring av hjernen));
- Røntgen av føtter og hender for å bestemme årsaken til fingrenes sammensmelting (dette er viktig for planlegging av senere kirurgiske inngrep);
- En konsultasjon med en medisinsk genetiker og en nevrokirurg kan bli foreskrevet.
Tester
Genetisk analyse av vanlige mutasjoner som forekommer i FGFR2-typegenet blir utført.
Differensiell diagnose
Dette syndromet må differensieres fra andre genetiske patologier der kraniosynostose observeres. Dette er sykdommer som Pfeiffer-, Crouzon-, Saethre-Chotzen- og Carpenter-syndromer. Molekylærgenetiske testmetoder brukes for å utelukke disse anomaliene.
Hvem skal kontakte?
Behandling Aper-syndromet
Kirurgi regnes som den eneste effektive behandlingen for Apert syndrom – det bidrar til å korrigere individuelle fysiske defekter og korrigerer også mental retardasjon.
Under denne prosedyren lukkes den koronale suturen for å forhindre mulig hjerneskade. Den vanligste teknikken er kraniofacial distraksjon, som involverer gradert skalletraksjon. Ortodontisk og/eller ortognatisk kirurgi utføres for å fjerne individuelle ansiktsdefekter.
I tillegg gjennomgår pasientene kirurgisk korreksjon av fingerfusjon.
Referanser
Medisinsk genetikk. Nasjonal guide. Redigert av Ginter EK, Puzyrev VP GEOTAR-Media. 2022