
Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
Martin-Bells syndrom
Medisinsk ekspert av artikkelen

Sist anmeldt: 04.07.2025
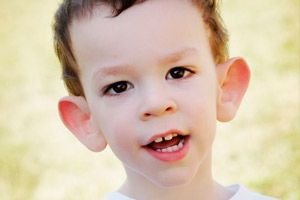
Martin-Bell syndrom ble beskrevet i 1943 av leger, som det ble oppkalt etter. Sykdommen er en genetisk lidelse som består av psykisk utviklingshemming. I 1969 ble endringer i kromosom X (skjørhet i den distale armen) som er karakteristiske for denne sykdommen identifisert. I 1991 oppdaget forskere genet som er ansvarlig for utviklingen av denne sykdommen. Denne sykdommen kalles også "skjørt X-syndrom". Både gutter og jenter er utsatt for sykdommen, men gutter er oftere (3 ganger) rammet.
Epidemiologi
Martin-Bell syndrom er en ganske vanlig sykdom: 0,3–1,0 av 1000 menn lider av denne sykdommen, og 0,2–0,6 av 1000 kvinner. Dessuten fødes barn med Martin-Bell syndrom på alle kontinenter med samme frekvens. Nasjonalitet, hudfarge, øyeform, levekår og folks velvære påvirker åpenbart ikke forekomsten av sykdommen. Hyppigheten av forekomsten er kun sammenlignbar med hyppigheten av Downs syndrom (1 sykdom av 600–800 nyfødte). En femtedel av mannlige bærere av det endrede genet er friske, har ingen kliniske eller genetiske abnormaliteter, resten har tegn på psykisk utviklingshemming fra mild til alvorlig form. Blant kvinnelige bærere er litt over en tredjedel syke.
Fragilt X-syndrom rammer omtrent 1 av 2500–4000 menn og 1 av 7000–8000 kvinner. Bæreprevalensen blant kvinner er anslått til å være 1 av 130–250; bæreprevalensen blant menn er anslått til å være 1 av 250–800.
Fører til Martin-Bells syndrom
Martin-Bell syndrom utvikler seg på grunn av fullstendig eller delvis opphør av kroppens produksjon av et spesifikt protein. Dette skjer på grunn av manglende respons fra FMR1-genet, lokalisert i X-kromosomet. Mutasjonen skjer som et resultat av omstrukturering av genet fra ustabile strukturelle varianter av gentilstandene (alleler), og ikke helt fra begynnelsen. Sykdommen overføres kun gjennom den mannlige linjen, og mannen er ikke nødvendigvis syk. Mannlige bærere overfører genet til døtrene sine i uendret form, slik at deres mentale retardasjon ikke er åpenbar. Ved videre overføring av genet fra mor til barna hennes, muterer genet, og alle tegnene som er karakteristiske for denne sykdommen, oppstår.
Patogenesen
Patogenesen til Martin-Bell syndrom er basert på mutasjoner i genapparatet, som fører til blokkering av produksjonen av FMR-protein, et protein som er livsviktig for kroppen, spesielt i nevroner, og som finnes i ulike vev. Forskning viser at FMR-proteiner er direkte involvert i prosessene med å regulere translasjoner som skjer i hjernevev. Fravær av dette proteinet eller kroppens begrensede produksjon av det fører til mental retardasjon.
I sykdommens patogenese regnes genhypermetylering som en nøkkellidelse, men det har ennå ikke vært mulig å definitivt identifisere mekanismen for utvikling av denne lidelsen.
Samtidig ble det også oppdaget locus heterogenitet i patologien, som er assosiert med polyallelisme, samt polylokus. Tilstedeværelsen av alleliske varianter av sykdomsutviklingen ble bestemt, som er forårsaket av eksistensen av punktmutasjoner, samt ødeleggelsen av FMRL-typegenet.
Pasientene har også to fragile tripletter som er følsomme for folsyre, lokalisert 300 kb, samt 1,5–2 millioner bp fra den fragile tripletten som inneholder FMR1-genet. Mekanismen for mutasjoner som forekommer i FRAXE- og FRAXF-genene (de er identifisert i de ovennevnte fragile triplettene) er relatert til mekanismen for lidelser ved Martin Bell syndrom. Denne mekanismen er forårsaket av spredning av GCC- og CGG-repetisjoner, som forårsaker metylering av de såkalte CpG-øyene. I tillegg til den klassiske formen for patologien, finnes det også to sjeldne typer som skiller seg på grunn av ekspansjonen av trinukleotidrepetisjoner (i mannlig og kvinnelig meiose).
Det ble funnet at pasienten i den klassiske formen av syndromet mangler et spesielt nukleocytoplasmatisk protein av typen FMR1, som utfører funksjonen med å binde forskjellige mRNA-er. I tillegg fremmer dette proteinet dannelsen av et kompleks som bidrar til å utføre translasjonsprosesser inne i ribosomer.
Symptomer Martin-Bells syndrom
Hvordan gjenkjenne sykdommen hos barn? Hva er de første tegnene? I de første månedene av et barns liv er det umulig å gjenkjenne Martin-Bell-symptomet, bortsett fra at det noen ganger observeres en reduksjon i muskeltonus. Etter et år er det kliniske bildet av sykdommen mer tydelig: barnet begynner å gå og snakke sent, noen ganger er talen helt fraværende. Han er hyperaktiv, vifter med armene tilfeldig, er redd for folkemengder og støy, er sta, det er skarpe sinneutbrudd, emosjonell ustabilitet, epileptiske anfall forekommer, får ikke øyekontakt. Hos pasienter med Martin-Bell syndrom avsløres sykdommen også av utseendet: ørene er utstående og store, pannen er tung, ansiktet er langstrakt, haken er utstående, strabismus, brede hender og føtter. De er også preget av endokrine forstyrrelser: ofte stor vekt, fedme, store testikler hos menn, tidlig pubertet.

Blant pasienter med Martin-Bell syndrom varierer intelligensnivået sterkt: fra mild psykisk utviklingshemming til alvorlige tilfeller. Hvis en normal person har en intelligenskvotient (IQ) på 100 i gjennomsnitt, og et geni har 130, har personer som er mottakelige for sykdommen 35–70.
Alle kliniske symptomer på patologien kan karakteriseres av en triade av hovedmanifestasjoner:
- oligofreni (IQ er 35-50);
- dysmorfofobi (utstående ører og prognatisme observeres);
- makroorkidisme, som oppstår etter pubertetens begynnelse.
Omtrent 80 % av pasientene har også bikuspidalklaffprolaps.
Imidlertid manifesterer den fulle formen av syndromet seg bare hos 60 % av alle pasienter. Hos 10 % oppdages kun psykisk utviklingshemming, og hos resten utvikler sykdommen seg med en annen kombinasjon av symptomer.
Blant de første tegnene på sykdommen, som oppstår i tidlig alder:
- det syke barnet viser betydelig psykisk utviklingshemming sammenlignet med andre jevnaldrendes utvikling;
- oppmerksomhets- og konsentrasjonsforstyrrelser;
- sterk stahet;
- barn begynner å gå og snakke ganske sent;
- hyperaktivitet og taleutviklingsforstyrrelser observeres;
- veldig sterke og ukontrollerbare anfall av sinne;
- mutisme kan utvikle seg - dette er en fullstendig mangel på tale hos et barn;
- babyen opplever sosial angst og er i stand til å få panikk på grunn av høy lyd eller andre høye lyder;
- barnet vifter ukontrollert og kaotisk med armene;
- sjenanse observeres, barnet er redd for å være på overfylte steder;
- fremveksten av ulike obsessive ideer, ustabil emosjonell tilstand;
- Babyen kan være motvillig til å få øyekontakt med folk.
Hos voksne observeres følgende symptomer på patologien:
- spesifikt utseende: et langstrakt ansikt med en tung panne, store utstående ører, en sterkt utstående hake;
- flate føtter, mellomørebetennelse og strabismus;
- puberteten skjer ganske tidlig;
- fedme kan utvikle seg;
- Ganske ofte observeres hjertefeil ved Martin-Bell syndrom;
- hos menn observeres forstørrelse av testiklene;
- leddenes artikulasjoner blir veldig mobile;
- vekt og høyde øker kraftig.
Diagnostikk Martin-Bells syndrom
For å diagnostisere Martin Bell syndrom må du kontakte en kvalifisert genetiker. Diagnosen stilles etter spesifikke genetiske tester som lar deg identifisere det defekte kromosomet.
Tester
I et tidlig stadium av sykdommen brukes en cytogenetisk metode, der et fragment av cellemateriale tas fra pasienten, som deretter tilsettes folsyre for å fremkalle endringer i kromosomene. Etter en viss tid identifiseres et område av kromosomet der det er en merkbar fortynning – dette er et tegn på tilstedeværelsen av fragilt X-syndrom.
Denne testen er imidlertid ikke egnet for diagnose i de senere stadiene av sykdommen, fordi nøyaktigheten reduseres av den utbredte bruken av multivitaminer som inneholder folsyre.
Integrert diagnostikk av Martin-Bell syndrom er en molekylærgenetisk undersøkelse, som består av å bestemme antallet såkalte trinukleotidrepetisjoner i genet.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Instrumentell diagnostikk
En svært spesifikk metode for instrumentell diagnostikk er PCR (polymerasekjedereaksjon), som lar en studere strukturen til aminosyrerester som finnes i X-kromosomet og dermed bestemme tilstedeværelsen av Martin Bell syndrom.
Det finnes også en egen, enda mer spesifikk metode for patologidiagnostikk – en kombinasjon av PCR og deteksjon ved bruk av kapillærelektroforese. Denne metoden er svært nøyaktig og oppdager kromosomal patologi hos pasienter med primær ovarialsvikt, samt ataksisk syndrom.
Tilstedeværelsen av defekten kan bestemmes etter å ha utført diagnostikk på EEG. Pasienter med denne sykdommen har lignende bioelektrisk hjerneaktivitet.
Hvilke tester er nødvendig?
Differensiell diagnose
Differensierte metoder som bidrar til å mistenke syndromet inkluderer:
- klinisk - 97,5 % av pasientene har tydelige tegn på psykisk utviklingshemming (moderat eller alvorlig); 62 % har store utstående ører; 68,4 % har stor, utstående hake og panne; 68,4 % av guttene har forstørrede testikler, 41,4 % har talevansker (ujevn talehastighet, ukontrollert volum osv.);
- cytogen - blod undersøkes for lymfocyttkultur, antall celler med fragilt X-kromosom per 100 studerte celler bestemmes;
- Elektroencefalografi - endringer i elektriske impulser i hjernen som er spesifikke for Martin-Bell syndrom registreres.
Hvem skal kontakte?
Behandling Martin-Bells syndrom
Ved behandling av voksne pasienter brukes antidepressiva med psykostimulerende midler. Medikamentell behandling overvåkes kontinuerlig av en psykolog og psykiater. I tillegg utføres mikroinjeksjonsprosedyrer med legemidler som Cerebrolysin (eller dets derivater), samt cytomediner (som Solcoseryl eller Lidase) i private klinikker.
Ved utvikling av ataksisk syndrom brukes blodfortynnende legemidler og nootropika. I tillegg foreskrives aminosyreblandinger og angioprotektorer. Kvinner med primær eggstokksvikt foreskrives korrigerende behandling med urtemedisiner og østrogener.
Glutaminreseptorantagonister brukes også i behandlingen.
Tradisjonelt innebærer behandlingen av Martin-Bell syndrom bruk av medisiner som påvirker symptomene på sykdommen, men ikke årsaken. Denne terapien innebærer forskrivning av antidepressiva, nevroleptika og psykostimulerende midler. Ikke alle legemidler er indisert for bruk hos barn, så listen over legemidler er ganske begrenset. Nevroleptika som kan brukes etter 3 år (den tidligste alderen for forskrivning) inkluderer haloperidol i dråper og tabletter, klorpromazin i løsning og periciazin i dråper. Dermed beregnes dosen av haloperidol for barn avhengig av kroppsvekt. For voksne foreskrives dosen individuelt. Det tas oralt, startende med 0,5–5 mg 2–3 ganger daglig, deretter økes dosen gradvis til 10–15 mg. Når forbedring oppstår, byttes de til en lavere dose for å opprettholde den oppnådde tilstanden. Ved psykomotorisk agitasjon foreskrives 5–10 mg intramuskulært eller intravenøst, flere repetisjoner er mulig etter 30–40 minutter. Den daglige dosen bør ikke overstige 100 mg. Bivirkninger i form av kvalme, oppkast, muskelspasmer, økt trykk, arytmi, etc. er mulige. Eldre personer bør ta spesielle forholdsregler, da tilfeller av plutselig hjertestans har blitt registrert, og tardiv dyskinesi (ufrivillige bevegelser) kan forekomme.
Antidepressiva øker aktiviteten i hjernestrukturer, lindrer depresjon, spenninger og forbedrer humøret. Disse legemidlene, som anbefales for bruk fra 5-8 år ved Martin-Bell syndrom, inkluderer klomipromin, sertralin, fluoksetin og fluvoksamin. Fluoksetin tas derfor oralt under måltider 1-2 ganger (helst i første halvdel av dagen), og starter med 20 mg per dag, økes til 80 mg om nødvendig. Eldre personer anbefales ikke en dose høyere enn 60 mg. Behandlingsforløpet bestemmes av legen, men ikke mer enn 5 uker.
Mulige bivirkninger: svimmelhet, angst, tinnitus, tap av appetitt, takykardi, ødem, etc. Forsiktighet er nødvendig ved forskrivning til eldre, personer med hjerte- og karsykdommer og diabetes.
Psykostimulerende midler er psykotrope legemidler som brukes til å forbedre oppfatningen av ytre stimuli: de skjerper hørsel, reaksjoner og syn.
Diazepam foreskrives som et beroligende middel mot nevroser, angst, epileptiske anfall og kramper. Det tas oralt, intravenøst, intramuskulært, rektalt (i endetarmen). Det foreskrives individuelt, avhengig av sykdommens alvorlighetsgrad, med de minste dosene på 5-10 mg, daglig - 5-20 mg. Behandlingsvarigheten er 2-3 måneder. For barn beregnes dosen under hensyntagen til kroppsvekt og individuelle egenskaper. Bivirkninger inkluderer sløvhet, apati, døsighet, kvalme, forstoppelse. Det er farlig å kombinere med alkohol, da avhengighet av stoffet er mulig.
Ved behandling av Martin-Bell syndrom har det vært tilfeller av bedring av tilstanden og med introduksjon av legemidler laget av animalsk materiale (hjerne): cerebrolysat, cerebrolysin, cerebrolysat-M. Hovedkomponentene i disse legemidlene er peptider som fremmer produksjonen av protein i nevroner, og dermed fyller på manglende protein. Cerebrolysin administreres som en stråle på 5-10 ml, behandlingsforløpet består av 20-30 injeksjoner. Legemidlet foreskrives til barn fra ett år, administrert intramuskulært hver dag 1-2 ml i en måned. Gjentatte økter med administrering er mulig. Bivirkninger i form av feber, kontraindisert for gravide kvinner.
Det ble gjort forsøk på å behandle sykdommen med folsyre, men bare det atferdsmessige aspektet ble bedre (nivået av aggresjon og hyperaktivitet sank, talen forbedret seg), og ingenting endret seg på det intellektuelle nivået. For å forbedre sykdomstilstanden foreskrives folsyre, fysioterapimetoder, logopedi, pedagogisk og sosial korreksjon er indisert.
Litiumpreparater anses også som effektive, ettersom de bidrar til å forbedre pasientens tilpasning til det sosiale miljøet, samt kognitiv aktivitet. I tillegg regulerer de også hans atferd i samfunnet.
Bruk av urter ved Martin-Bell syndrom er mulig som antidepressiva. Urter som bidrar til å lindre spenninger, angst og forbedre søvn inkluderer valerian, peppermynte, timian, johannesurt og kamille. Infusjoner tilberedes som følger: til 1 teskje tørre urter trenger du et glass kokende vann, avkok trekkes i minst 20 minutter, tas hovedsakelig om kvelden før leggetid eller om ettermiddagen. En skje med honning ville være et godt tillegg til dem.
Fysioterapibehandling
For å eliminere nevrologiske manifestasjoner utføres spesielle fysioterapeutiske prosedyrer - som øvelser i bassenget, muskelavslapning og akupunktur.
Kirurgisk behandling
Et viktig stadium i behandlingen anses også å være plastikkirurgiske metoder - operasjoner som bidrar til å forbedre pasientens utseende. Plastisk kirurgi av lemmer og ørene utføres, samt kjønnsorganene. Korrigering av gynekomasti med epispadi utføres også, samt andre defekter i utseende.
Forebygging
Den eneste metoden for å forebygge sykdommen er prenatal screening av gravide. Det finnes spesielle undersøkelser som muliggjør tidlig oppdagelse av patologien, hvoretter det anbefales å avslutte svangerskapet. Alternativt brukes IVF, som kan hjelpe barnet med å arve et sunt X-kromosom.
Forebygging av pasienten avhenger av om genmutasjonen har oppstått igjen eller er arvet. For dette utføres molekylærgenetisk diagnostikk. Det faktum at testen ikke avdekket det "skjøre X-kromosomet" hos slektninger taler for mutasjonens "ferskhet", noe som betyr at risikoen for å få et barn med Martin-Bell syndrom er svært liten. I familier der det er syke mennesker, vil testen bidra til å unngå gjentatte tilfeller.
Prognose
Prognosen for Martin-Bell syndrom er gunstig for livet, men ikke for bedring. Forventet levealder avhenger av alvorlighetsgraden av sykdommen og tilhørende defekter. Pasienten kan leve et normalt liv. Ved alvorlige former for Martin-Bell syndrom risikerer pasientene livslang uførhet.
Forventet levealder
Martin Bell syndrom har ingen alvorlig negativ innvirkning på helsen, så forventet levealder for de fleste som har blitt diagnostisert med denne patologien, avviker ikke fra standardindikatorer.