
Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
Akondroplasi
Medisinsk ekspert av artikkelen

Sist anmeldt: 12.07.2025
Det finnes mange sjeldne medfødte sykdommer, og en av dem er et brudd på beinvekst - achondroplasi, som fører til alvorlig uforholdsmessig kort statur.
I avsnittet om utviklingsanomalier i ICD-10 er koden for denne typen arvelig osteokondral dysplasi med vekstdefekter i rørformede bein og ryggrad Q77.4 [ 1 ]
Epidemiologi
Når det gjelder forekomsten av akondroplasi, er statistiske data fra ulike studier tvetydige. Noen hevder at denne anomalien forekommer hos én nyfødt av 10 000, andre - hos én av 26–28 000, og atter andre - 4–15 tilfeller av 100 000. [ 2 ]
Det finnes også informasjon om at når faren er over 50 år gammel, er forekomsten av akondroplasi hos barn ett tilfelle per 1875 nyfødte.
Fører til akondroplasi
Årsaken til akondroplasi er et brudd på osteogenesen, spesielt en av typene intrauterin ossifikasjon av diafysene i skjelettets rørformede bein - endokondral ossifikasjon, der brusk modifiseres til beinvev. For mer informasjon, se - Beinutvikling og vekst
Forstyrrelse av ossifikasjonen av lange bein, dvs. føtal akondroplasi, oppstår på grunn av mutasjoner i membrantyrosinkinasegenet - fibroblastvekstfaktorreseptor 3 (FGFR3 på kromosom 4p16.3), som påvirker cellevekst og -differensiering. Tilstedeværelsen av FGFR3-mutasjoner er assosiert med genetisk ustabilitet og endringer i antall kromosomer (aneuploidi).
Akondroplasi overføres til et barn som et autosomalt dominant trekk, det vil si at han mottar én kopi av det mutante genet (som er dominant) og ett normalt gen på et par ikke-kjønnsmessige (autosomale) kromosomer. Dermed er arvetypen for denne defekten autosomalt dominant, og anomalien kan manifestere seg hos 50 % av avkommet når en kombinasjon av alleler av dette genet (genotypen) krysses.
I tillegg kan mutasjoner være sporadiske, og som praksis viser, blir barn med akondroplasi i 80 % av tilfellene født av foreldre med normal høyde.
Risikofaktorer
De viktigste risikofaktorene for fødsel av barn med akondroplasi er arvelige. Hvis en av foreldrene har denne defekten, er sannsynligheten for å få et sykt barn estimert til 50 %; hvis begge foreldrene har denne anomalien, er den også 50 %, men med en 25 % risiko for homozygot akondroplasi, som fører til død før fødselen eller i tidlig spedbarnsalder.
Med farens alder (nærmere 40 år og eldre) øker risikoen for en ny mutasjon (de novo-mutasjon) av FGFR3-genet.
Patogenesen
Eksperter forklarer patogenesen til akondroplasi og understreker viktigheten av transmembranproteinet tyrosinproteinkinase (kodet av FGFR3-genet) i reguleringen av deling, differensiering og apoptose av celler i bruskvev i vekstplatene - kondrocytter, samt normal utvikling av skjelettet - osteogenese og mineralisering av beinvev.
Under embryonal utvikling, i nærvær av en genmutasjon, blir reseptorene for fibroblastvekstfaktor 3 mer aktive. Økningen i deres funksjoner forstyrrer overføringen av cellulære signaler og samspillet mellom den ekstracellulære delen av dette proteinet og polypeptidfibroblastvekstfaktorer (FGF). Som et resultat oppstår en svikt: proliferasjonsstadiet av bruskceller blir kortere, og differensieringen deres begynner tidligere enn forventet. Alt dette fører til feil dannelse og fusjon av hodeskallebein og skjelettdysplasi - en reduksjon i lange bein, som er ledsaget av uttalt kortvoksthet eller dvergvekst.
Og to tredjedeler av tilfellene av dvergvekst er assosiert med akondroplasi.
Symptomer akondroplasi
Unormal beinvekst forårsaker kliniske symptomer på akondroplasi som:
- uttalt kort statur (uforholdsmessig dvergvekst) med en gjennomsnittlig voksenhøyde på 123-134 cm;
- forkortelse av de proksimale delene av nedre og øvre lemmer med en relativt normal torsostørrelse;
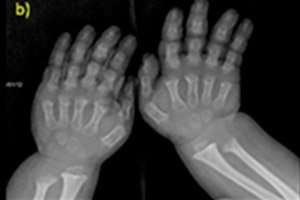
- forkortede fingre og tær;
- forstørret hode (makro- eller megalocephaly); [ 3 ]
- spesifikke ansiktstrekk i form av en utstående panne og hypoplasi av den midtre delen av ansiktet - en nedsunket neserygg.
- smal kraniocervikal overgang. Noen spedbarn med akondroplasi dør i løpet av det første leveåret av komplikasjoner relatert til kraniocervikal overgang. Populasjonsstudier tyder på at denne økte dødsrisikoen kan være så høy som 7,5 % uten evaluering og intervensjon.[ 4 ]
- Dysfunksjon i mellomøret er ofte et problem [ 5 ], og hvis den ikke behandles riktig, kan den føre til konduktivt hørselstap som er alvorlig nok til å forstyrre taleutviklingen. Mer enn halvparten av barna vil trenge en trykkutjevningsslange. [ 6 ] Totalt sett har omtrent 40 % av personer med akondroplasi funksjonelt signifikant hørselstap. Utviklingen av ekspressivt språk er også ofte forsinket, selv om det er tvilsomt hvor sterkt forholdet mellom hørselstap og problemer med ekspressivt språk er.
- Bøying av tibia er svært vanlig hos personer med akondroplasi. Over 90 % av ubehandlede voksne har en viss grad av bøying.[ 7 ] «Bøying» er faktisk en kompleks deformitet som følge av en kombinasjon av lateral tilt, intern torsjon av tibia og dynamisk ustabilitet i kneet.[ 8 ]
Spedbarn med akondroplasi kjennetegnes av muskulær hypotoni, som gjør at de begynner å lære bevegelsesferdigheter og gå senere. Intelligens og kognitive evner påvirkes ikke av denne utviklingsdefekten. [ 9 ], [ 10 ]
Konsekvenser og komplikasjoner
Denne typen arvelig osteokondral dysplasi er preget av følgende komplikasjoner og konsekvenser:
- tilbakevendende ørebetennelser;
- obstruktiv søvnapné;
- vannhode;
- malokklusjon og skjeve tenner:
- deformasjon av bena (varus eller valgus) med endring i gangart;
- hypertrofiert lordose i korsryggen eller dens krumning (thoracolumbal kyfose eller lumbal skoliose) - med ryggsmerter når man går;
- leddsmerter (på grunn av feil plassering av bein eller kompresjon av nerverøtter);
- Spinalstenose og ryggmargskompresjon; Den vanligste medisinske plagen i voksen alder er symptomatisk spinalstenose som involverer L1-L4. Symptomene varierer fra intermitterende, reversibel claudicatio indusert av trening til alvorlig, irreversibel bendysfunksjon og urinretensjon.[ 11 ] Claudicatio og stenose kan forårsake både sensoriske (nummenhet, smerte, tyngde) og motoriske symptomer (svakhet, snubling, begrenset gangutholdenhet). Vaskulær claudicatio skyldes hevelse i blodårene etter å ha stått og gått, og er fullstendig reversibel ved hvile. Spinalstenose er den faktiske lesjonen i ryggmargen eller nerveroten av det stenotiske beinet i spinalkanalen, og symptomene er irreversible. Symptomer lokalisert til et bestemt dermatom kan skyldes stenose av spesifikke nerverotforamina.
- en reduksjon i brystveggen med begrenset lungevekst og redusert lungefunksjon (alvorlig kortpustethet). I spedbarnsalderen har en liten gruppe personer med akondroplasi restriktive lungeproblemer. Små bryster og økt brystkomfort resulterer i redusert lungekapasitet og restriktiv lungesykdom [ 12 ]
Andre ortopediske problemer
- Leddsvakhet. De fleste ledd er hypermobile i barndommen. Vanligvis har dette liten effekt, bortsett fra kneustabilitet hos noen.
- Discoid lateral menisk: Denne nylig identifiserte strukturelle abnormaliteten kan føre til kroniske knesmerter hos noen.[ 13 ]
- Artritt: Konstitutiv aktivering av FGFR-3, som ved akondroplasi, kan beskytte mot utvikling av artritt.[ 14 ]
- Acanthosis nigricans ses hos omtrent 10 % av personer med akondroplasi.[ 15 ] I denne populasjonen gjenspeiler det ikke hyperinsulinemi eller malignitet.
Homozygot akondroplasi forårsaket av bialleliske patogene varianter ved nukleotid 1138 av FGFR3 er en alvorlig lidelse med radiologiske funn som er kvalitativt forskjellige fra de som sees ved akondroplasi. Tidlig død skyldes respirasjonssvikt på grunn av en liten brystvegg og nevrologiske underskudd på grunn av cervikomedullær stenose [Hall 1988].
Diagnostikk akondroplasi
Hos de fleste pasienter stilles diagnosen akondroplasi basert på karakteristiske kliniske tegn og radiografiske funn. Hos spedbarn eller i fravær av noen symptomer brukes genetisk testing, som karyotypeanalyse, for å stille en endelig diagnose.[16 ]
Ved utførelse av prenatal diagnostikk ved bruk av molekylærgenetisk metode kan analyser av fostervann eller en chorionvillusprøve utføres.
Tegn på akondroplasi på ultralyd av fosteret - forkortelse av lemmer og typiske ansiktstrekk - visualiseres etter 22 ukers svangerskap.
Instrumentell diagnostikk inkluderer også røntgen av skjelettet eller ultralyd av beinene. Og røntgen bekrefter diagnosen basert på data som en stor hodeskalle med en smal occipital foramen og en relativt liten base; korte rørformede bein og forkortede ribbein; korte og flate virvellegemer; innsnevret spinalkanal, redusert størrelse på iliacvingene.
Differensiell diagnose
Differensialdiagnostikk ved hypofysedvergvekst, medfødt spondyloepifyseal og diastrofisk dysplasi, hypokondroplasi, Shereshevsky-Turner og Noonan syndromer, pseudoakondroplasi er nødvendig. Forskjellen mellom pseudoakondroplasi og akondroplasi er derfor at hos pasienter med dvergvekst ved pseudoakondroplasi er hodestørrelsen og ansiktstrekkene normale.
Hvem skal kontakte?
Behandling akondroplasi
Anbefalinger for behandling av barn med akondroplasi er utarbeidet av American Academy of Pediatrics Committee on Genetics. Disse anbefalingene er ment å gi veiledning og er ikke ment å erstatte individuell beslutningstaking. En nylig gjennomgang [Pauli & Botto 2020] inneholder også retningslinjer. Det finnes spesialklinikker som spesialiserer seg i behandling av skjelettdysplasi; anbefalingene deres kan avvike noe fra disse generelle anbefalingene.
Anbefalingene inkluderer (men er ikke begrenset til) følgende.
Hydrocephalus. Hvis tegn eller symptomer på økt intrakranielt trykk oppstår (f.eks. akselerert hodevekst, vedvarende utbulende fontanell, merkbar forstørrelse av overfladiske vener i ansiktet, irritabilitet, oppkast, synsforandringer, hodepine), er henvisning til nevrokirurg nødvendig.
Den antatte etiologien for hydrocephalus ved akondroplasi er økt intrakranielt venetrykk på grunn av stenose av jugularforamina. Derfor har standardbehandlingen vært ventrikuloperitoneal shunting. Endoskopisk tredje ventrikulostomi kan imidlertid være gunstig hos noen individer,[ 17 ] noe som antyder at andre mekanismer, som obstruksjon av fjerde ventrikkelutløp på grunn av kraniocervikal stenose, kan være involvert.[ 18 ]
Kraniocervikal overgangstenose. Beste prediktorer for behov for suboccipital dekompresjon:
- Hyperrefleksi eller klonus i nedre ekstremiteter
- Sentral hypopné ved polysomnografi
- Reduksjon i foramen magnum-størrelse bestemt ved computertomografi av kraniocervikal overgang og sammenlignet med normer for barn med akondroplasi.[ 19 ]
- Bevis for ryggmargskompresjon og/eller T2-vektede signalavvik har nylig blitt foreslått som en annen faktor å vurdere når man bestemmer seg for å operere.
Hvis det er tydelige tegn på symptomatisk kompresjon, bør det umiddelbart henvises til en pediatrisk nevrokirurg for dekompresjonskirurgi. [ 20 ]
Behandling av obstruktiv søvnapné kan omfatte:
- Adenotonsillektomi
- Positivt luftveistrykk
- Trakeostomi i ekstreme tilfeller
- Vekttap
Disse tiltakene kan føre til forbedring av søvnforstyrrelser og noe forbedring av nevrologisk funksjon.[ 21 ]
I sjeldne tilfeller der obstruksjonen er alvorlig nok til å kreve trakeostomi, har kirurgisk fremføring av midtflaten blitt brukt for å lindre obstruksjon i øvre luftveier.[ 22 ]
Dysfunksjon i mellomøret. Hyppige mellomøreinfeksjoner, vedvarende væske i mellomøret og påfølgende hørselstap bør behandles aggressivt ved behov. Langtidsbruk av sonde anbefales fordi de ofte er nødvendige frem til syv eller åtte år.[ 23 ]
Når problemer oppstår i alle aldre, anbefales det å bruke passende behandlingsmetoder.
Kortvoksthet. Flere studier har evaluert veksthormonbehandling (GH) som en mulig behandling for akondroplasi ved kortvoksthet.[ 24 ]
Samlet sett viser disse og andre serier en innledende akselerasjon av veksten, men effekten avtar over tid.
I gjennomsnitt kan du forvente en økning i voksenhøyde på bare omtrent 3 cm.
Forlengelse av lemmer ved bruk av ulike teknikker er fortsatt et alternativ for noen. Høydeøkning på opptil 30–35 cm kan oppnås. [ 25 ] Komplikasjoner er vanlige og kan være alvorlige.
Mens noen anbefaler å utføre disse prosedyrene så tidlig som seks til åtte år, anbefaler mange barneleger, kliniske genetikere og etikere å utsette slike operasjoner til en ung person er i stand til å delta i å ta en informert beslutning.
I hvert fall i Nord-Amerika velger bare en liten andel av de berørte individene å gjennomgå avansert lemforlengelse. Little People of America Medical Advisory Board har gitt ut en uttalelse angående bruk av avansert lemforlengelse.
Fedme: Tiltak for å forebygge fedme bør starte i tidlig barndom. Standardbehandlinger for fedme bør være effektive hos personer med akondroplasi, selv om kaloribehovet er lavere. [ 26 ]
Standard vekt- og vekt-for-høyde-diagrammer spesifikke for akondroplasi bør brukes til å spore fremgang. Det er viktig å merke seg at disse kurvene ikke er perfekte vekt-for-høyde-kurver; de ble utledet fra tusenvis av datapunkter fra personer med akondroplasi.
Standarder for kroppsmasseindeks (BMI) ble utviklet for barn i alderen 16 år og yngre. [ 27 ] BMI er ikke standardisert for voksne med akondroplasi; sammenligninger med BMI-kurver for gjennomsnittlig høyde vil gi misvisende resultater. [ 28 ]
Varusdeformitet. Årlig ortopedisk oppfølging av enten en lege med kjennskap til akondroplasi eller en ortopedisk kirurg anbefales. Kriterier for kirurgisk inngrep er publisert.[ 29 ]
Tilstedeværelse av progressiv symptomatisk kurve krever henvisning til en ortoped. Asymptomatisk varusdeformitet i seg selv krever vanligvis ikke kirurgisk korreksjon. Ulike tiltak kan velges (f.eks. styrt vekst ved bruk av åtte plater, valgusosteotomi og derotasjonsosteotomi). Det finnes ingen kontrollerte studier som sammenligner resultatene av behandlingsalternativene.
Kyfose. Spedbarn med akondroplasi utvikler ofte en fleksibel kyfose. Det finnes en protokoll for å forhindre utvikling av en fast vinkelkyfose, som inkluderer å unngå fleksible barnevogner, husker og bæreseler. Råd mot ustøttet sitting; bruk alltid mottrykk på ryggen når du holder babyen.
- Kyfose bedres betydelig eller forsvinner hos de fleste barn etter at de har inntatt en ortograd stilling og begynt å gå. [ 30 ]
- Hos barn som ikke spontant går i remisjon etter økt kroppsstyrke og begynt å gå, er det vanligvis tilstrekkelig med avstivning for å forhindre vedvarende thorakolumbal kyfose.[ 31 ]
- Hvis alvorlig kyfose vedvarer, kan det være nødvendig med ryggkirurgi for å forhindre nevrologiske komplikasjoner.[ 32 ]
Spinalstenose: Hvis alvorlige tegn og/eller symptomer på spinalstenose oppstår, er det nødvendig med øyeblikkelig henvisning til en kirurgisk spesialist.
Utvidet og bred laminektomi anbefales vanligvis. Relevansen av prosedyren avhenger av nivået (f.eks. thorax eller lumbal) og graden av stenose. Pasientene hadde bedre resultater og forbedret funksjon jo raskere de ble operert etter symptomdebut [ 33 ].
Vaksinasjoner: Ingenting om akondroplasi utelukker all rutinemessig vaksinasjon. Gitt den økte respiratoriske risikoen er DTaP-, pneumokokk- og influensavaksiner spesielt viktige.
Tilpasningsbehov: På grunn av lav vekst kan det være nødvendig med tilpasninger i omgivelsene. På skolen kan dette inkludere krakker, senkede lysbrytere, toaletter i passende høyde eller andre tilgjengelige midler, lavere pulter og fotstøtter foran stoler. Alle barn skal kunne forlate bygningen selvstendig i tilfelle en nødsituasjon. Små hender og svake sener kan gjøre finmotorikk vanskelig. Passende tilrettelegging inkluderer bruk av et mindre tastatur, vektpenner og glattere skriveflater. De fleste barn bør ha en IOP eller en 504-plan.
Pedalforlengere er nesten alltid nødvendige for sykling. Modifikasjoner av arbeidsstasjoner som lavere bord, mindre tastaturer, trinn og toaletttilgang kan også være nødvendig.
Sosialisering: På grunn av den svært merkbare korte veksten forbundet med akondroplasi, kan berørte individer og deres familier ha problemer med å sosialisere og tilpasse seg skolen.
Støttegrupper som Little People of America, Inc (LPA) kan hjelpe familier med å håndtere disse problemene gjennom støtte fra jevnaldrende, personlig eksempel og sosiale bevissthetsprogrammer.
Informasjon om sysselsetting, utdanning, rettigheter for funksjonshemmede, adopsjon av lave barn, medisinske problemer, passende klær, tilpasningsmidler og foreldrerollen er tilgjengelig gjennom et nasjonalt nyhetsbrev, seminarer og workshops.
Det finnes ingen medisin eller ikke-medikamentell behandling som kan kurere denne medfødte defekten.
Fysioterapi er den vanligste behandlingen; behandling kan også være nødvendig for hydrocephalus (ved shunt eller endoskopisk ventrikulostomi), fedme, [ 34 ] apné, [ 35 ] mellomørebetennelse eller spinalstenose.
På noen klinikker, etter at barnet har fylt fem til syv år, gjennomfører de kirurgisk behandling: forlengelse av bein i leggene, lårene og til og med skuldrene eller korrigering av deformiteten – ved hjelp av operasjoner og spesielle ortopediske apparater – i tre til fire trinn, som hver varer i opptil 6–12 måneder.
Terapi under utredning
Administrasjon av en C-type natriuretisk peptidanalog gjennomgår kliniske studier. Innledende resultater har vist at den tolereres godt og resulterer i en økning i veksthastighet fra baseline hos barn med akondroplasi ( studiested ). [ 36 ] Konjugert C-type natriuretisk peptid gjennomgår også for tiden kliniske studier. [ 37 ] Andre hensyn inkluderer tyrosinkinasehemming [ 38 ], meklizin [ 39 ] og en løselig rekombinant human FGFR3-lokkekur. [ 40 ]
Søk på clinicaltrials.gov i USA og EUs register over kliniske studier i Europa for informasjon om kliniske studier for et bredt spekter av sykdommer og tilstander.
Forebygging
Det eneste forebyggende tiltaket er prenatal diagnostikk av medfødte sykdommer. [ 41 ], [ 42 ]
Prognose
Hvor lenge lever personer med akondroplasi? Omtrent 10 år kortere enn gjennomsnittlig forventet levealder.
Siden patologiske forandringer i beinvev og ledd fører til begrensninger i egenomsorg og mobilitet, får barn med denne diagnosen status som funksjonshemmede. På lang sikt har de fleste pasienter en normal prognose, men med alderen er det økt risiko for hjertesykdom. [ 43 ]