
Alt iLive-innhold blir gjennomgått med medisin eller faktisk kontrollert for å sikre så mye faktuell nøyaktighet som mulig.
Vi har strenge retningslinjer for innkjøp og kun kobling til anerkjente medieområder, akademiske forskningsinstitusjoner og, når det er mulig, medisinsk peer-evaluerte studier. Merk at tallene i parenteser ([1], [2], etc.) er klikkbare koblinger til disse studiene.
Hvis du føler at noe av innholdet vårt er unøyaktig, utdatert eller ellers tvilsomt, velg det og trykk Ctrl + Enter.
Alkaptonuri er en medfødt enzymavvik
Medisinsk ekspert av artikkelen

Sist anmeldt: 04.07.2025
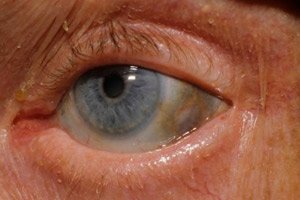
En av de svært sjeldne metabolske lidelsene, alkaptonuri, refererer til medfødte anomalier i metabolismen av aminosyren tyrosin.
Dette syndromet kan også kalles homogentisatoksidasemangel, homogentisinuri, arvelig ochronose eller svarturinsykdom.[ 1 ]
Epidemiologi
Ifølge statistikk er det ikke mer enn ni tilfeller av alkaptonuri per 1 million mennesker. Og i de fleste europeiske land er det ett tilfelle per 100–250 tusen levendefødte.
Blant europeiske land er unntaket Slovakia (spesielt den relativt lille nordvestlige regionen), hvor forekomsten av alkaptonuri er ett tilfelle per 19 000 nyfødte. Dette skyldes mest sannsynlig at blant de slovakiske romfamiliene som bor der, er nivået av innavl (ekteskap mellom søskenbarn) det høyeste i Europa: 10–14 %. [ 2 ]
Fører til alkaptonuri
De eksakte årsakene til alkaptonuri, som en medfødt lidelse i katabolismen (metabolsk nedbrytning) av den aromatiske (homosykliske) α-aminosyren tyrosin, er fastslått: denne typen metabolsk lidelse er en konsekvens av homozygote eller sammensatte heterozygote mutasjoner av et av de tusenvis av genene på kromosom 3, mer presist, HGD-genet på lokus 3q21-q23 på kromosomets lange arm. Dette genet koder for nukleotidsekvensene til leverenzymet homogentisat-1,2-dioksygenase [ 3 ] (også kalt homogentisinsyreoksidase eller homogentisatoksidase) - et jernholdig metalloprotein som er nødvendig for et av stadiene av tyrosin-nedbrytning i kroppen. [ 4 ], [ 5 ]
Dermed er alkaptonuri en defekt i enzymet homogentisat-1,2-dioksygenase, eller mer presist, et resultat av genetisk betinget mangel eller fullstendig fravær. [ 6 ]
Siden alkaptonuri er en medfødt enzymmangel, arves det autosomalt recessivt. Det vil si at for at alkaptonuri skal forekomme hos barn, må begge foreldrene ha et modifisert gen for enzymet, siden hver av dem bare overfører én kopi av genet av de to tilgjengelige til barnet.
I følge de nyeste dataene finnes det mer enn to hundre varianter av modifikasjon av HGD-genet, og missense-mutasjoner, translokasjon og spleising observeres oftest.
Risikofaktorer
Den eneste risikofaktoren for å utvikle denne medfødte enzymopatien er dens tilstedeværelse i familiehistorien og arv av to modifiserte kopier av HGD-genet, dersom foreldrene ikke har alkaptonuri (risikoen for å overføre anomalien er 25 %), eller en av foreldrene har denne lidelsen. [ 7 ]
Patogenesen
Tyrosin spiller en nøkkelrolle i syntesen av proteiner, produksjonen av kromoproteiner – hudpigmentet melanin, samt skjoldbruskkjertelhormoner og katekolamin-nevrotransmittere.
Mekanismen for å regulere mengden tyrosin i celler er svært kompleks, og kroppen normaliserer overskuddsinnholdet ved å bryte det ned. Prosessen med tyrosinkatabolisme, som alle aromatiske aminosyrer, er flertrinns og skjer i flere stadier. Hvert trinn i den metabolske nedbrytningen av tyrosin skjer med deltakelse av et spesifikt enzym og dannelsen av en mellomliggende forbindelse.
Så først brytes aminosyren ned til para-hydroksyfenylpyruvat, som omdannes til alkapton – 2,5-dihydroksyfenyleddiksyre eller homogentisinsyre. Deretter skal alkaptonet omdannes til maleeddiksyre, men dette skjer ikke. [ 8 ]
Og patogenesen til alkaptonuri består av opphør av biokjemiske reaksjoner av tyrosinkatabolisme i dannelsesstadiet av homogentisinsyre: det er rett og slett ikke noe enzym som trengs for å bryte det ned – homogentisatoksidase.
Homogentisinsyre brukes ikke av kroppen og kan akkumuleres ved utskillelse gjennom nyrene. I tillegg oksideres den til benzokinoacetat (benzokinoneddiksyre), som ved å binde seg til molekyler i vev og kroppsvæsker danner biopolymerforbindelser farget som melanin.
Opphopning av disse mellomproduktene i vevet fører til en forstyrrelse av bruskvevets kollagenstruktur, noe som reduserer elastisiteten – med forekomst av mange kliniske tegn på alkaptonuri og utvikling av komplikasjoner.
Symptomer alkaptonuri
Alkaptonuri hos nyfødte og spedbarn kjennetegnes av mørkfarging av urinen. Når urinen på bleier, skjørt og undertøy utsettes for luft, blir den mørkebrun. Dette skyldes akkumulering og frigjøring av homogentisinsyre, som oksideres til benzokinoacetat. [ 9 ]
I mangel av andre symptomer blir alkaptonuri hos små barn ofte ikke oppdaget i tide, siden urinen kan bli mørkere etter flere timers vannlating. Ifølge noen data blir bare en femtedel av barn under 12 måneder som er født med denne enzymmangelen, identifisert i kliniske settinger. Derfor er det svært viktig at foreldre er oppmerksomme på å ta vare på spedbarnene sine.
I tillegg inkluderer tidlige tegn pigmentering (blågrå farge) av senehinnene i øynene og brusken i ørene og nesen, noe som ofte kalles ochronose.[ 10 ]
Over tid dukker det opp andre symptomer:
- alvorlig pigmentering av huden på kinnbein, armhuler og kjønnsorganer;
- flekker på klær ved kontakt med svette områder på kroppen;
- anfall av generell svakhet;
- hes stemme.
Det bør huskes at alkaptonuri og ochronose, som nevnt ovenfor, er synonyme navn for den samme lidelsen i tyrosinkatabolisme.
Lønnesirup-urinsykdom og alkaptonuri. Medfødt lønnesirup-urinsykdom eller leucinose er også en metabolsk lidelse, har samme arvemønster, og til og med mutasjoner forekommer på samme kromosom, men påvirker genet som koder for enzymkomplekset av forgrenet α-keto-syredehydrogenase. På grunn av dette kan ikke kroppen bryte ned visse komponenter av proteiner, spesielt aminosyrene leucin, isoleucin og valin. Ved denne sykdommen har urin (og ørevoks) en søt lukt; i tillegg inkluderer det kliniske bildet av denne typen organisk acidemi hypopigmentering, svingninger i blodtrykk, anfall, oppkast og diaré, et fall i blodsukkernivået, ketoacidose, hallusinasjoner, etc. Dødeligheten hos barn er ganske høy; hos voksne kan koma og død oppstå uten behandling på grunn av hjerneødem.
Albinisme og alkaptonuri er kun «forent» av tyrosin. Albinisme, inkludert okulokutan, er forårsaket av genetiske mutasjoner som påvirker produksjonen av pigmentet melanin. Medfødte forandringer er observert i TYR-genet på kromosom 11 (11q14.3), som koder for tyrosinase, et kobberholdig melanosomenzym som er nødvendig for dannelsen av hudpigment basert på tyrosinmetabolismeprodukter. Denne sykdommen er mye vanligere enn alkaptonuri.
Komplikasjoner og konsekvenser
Forårsaket av virkningen av mellomliggende metabolitter av tyrosin – homogentisinsyre og benzokinoneddiksyre – oppstår konsekvensene og komplikasjonene av alkaptonuri på grunn av avsetning av reaktive pigmenterte polymerer, ødeleggelse av kollagenfibriller og forverring av bruskens tilstand (med en reduksjon i deres motstand mot mekanisk stress).
Med årene, i voksen alder, utvikles degenerativ artritt og slitasjegikt i store ledd (hofte, korsbein og kne); mellomvirvelrommene blir smalere (spesielt i korsryggen og brystryggen) – med forkalkning og dannelse av osteofytter; tettheten av vevet i de subkondrale beinplatene avtar, og de underliggende beinene kan gjennomgå patologisk ombygging med dannelse av utvekster og deformasjon. [ 11 ]
Skade på hjerteklaffene (aorta- og mitralklaffene) og koronararteriene kan observeres – med tegn på koronar hjertesykdom, samt dannelse av steiner i nyrene og prostata – på grunn av den samme forkalkningen. [ 12 ], [ 13 ]
Diagnostikk alkaptonuri
Diagnosen medfødte metabolske forstyrrelser stilles vanligvis på grunnlag av studiet av kroppens biologiske væsker.
Basert på hvilke tester og reaksjoner kan alkaptonuri diagnostiseres? Urinprøver er nødvendige for å oppdage homogentisinsyre og bestemme nivået (normalt – 20–30 mg per dag, forhøyet – 3–8 g). En urinprøve undersøkes ved gasskromatografi eller massespektrometri, ved bruk av væskekromatografi; en screeningtest for tilstedeværelse av jernklorid i urinen er mulig. [ 14 ]
Det finnes også en metode for rask diagnostikk – bestemmelse av alkapton i tørkede urinflekker på papir (etter fargeintensitet).
Ved avklaring av diagnosen innebærer instrumentell diagnostikk (radiografi) å identifisere radiologiske tegn på slitasjegikt og andre leddpatologier hos pasienter.
Diagnosen bekreftes ved hjelp av molekylærgenetiske metoder for å diagnostisere arvelige sykdommer, som genetisk testing og DNA-sekvensering. [ 15 ]
Differensiell diagnose
Differensialdiagnose inkluderer hemokromatose og akutt leversvikt hos nyfødte, melaninuri, akutt intermitterende porfyri, hemofagocytisk lymfohistiocytose, primær mitokondriell patologi, revmatoid artritt, Bekhterevs sykdom.
Hvem skal kontakte?
Behandling alkaptonuri
Hovedbehandlingen for alkaptonuri er oral administrering av store doser (minst 1000 mg per dag) askorbinsyre. Hos barn øker dette utskillelsen av homogentisinsyre i urinen, og hos voksne reduserer det urininnholdet av derivatet benzokinoneddiksyre, og bremser bindingen til bindevevsstrukturer i leddene og kollagen. [ 16 ]
Vesteuropeiske klinikker tester legemidlet Nitisinone (Orfalin), et legemiddel fra gruppen metabolitter som hemmer den andre fasen av tyrosinkatabolisme: omdannelsen av para-hydroksyfenylpyruvat til homogentisinsyre. Bruk av dette farmakologiske midlet fører imidlertid til akkumulering av tyrosin og kan forårsake alvorlige bivirkninger, inkludert hornhinneuklarhet og fotofobi, neseblod og mageblødning, leversvikt, endringer i blodet, etc. Likevel er Nitisinone FDA-godkjent i USA for behandling av type I tyrosinemi. [17 ], [ 18 ]
Derfor utføres fysioterapibehandling – treningsterapi for å øke muskelstyrken og forbedre leddmobiliteten, balneoterapi og peloidbehandling for å begrense smerte – for leddproblemer forårsaket av alkaptonuri.
Selv om tyrosin ikke bare tilføres fra mat, men også produseres i kroppen, anbefales det at pasienter med alkaptonuri følger et proteinfattig kosthold og begrenser inntaket av matvarer som er rike på tyrosin, først og fremst storfekjøtt og svinekjøtt, meieriprodukter (spesielt oster), belgfrukter, nøtter og frø.
Forebygging
Det er umulig å forebygge genmutasjoner, men for å forhindre fødsel av barn med høy risiko for medfødte lidelser, finnes det medisinsk genetisk rådgivning, som er nødvendig før et planlagt svangerskap for de parene hvis familiehistorie inkluderer arvelige sykdommer. [ 19 ]
Prognose
Dødelig utgang fra alkaptonuri er svært sjeldne, og døden kan være forårsaket av alvorlige komplikasjoner som involverer hjertet og nyrene. Så den totale forventede levealderen for personer med alkaptonuri er god.
Men livskvaliteten reduseres på grunn av intense smerter i ledd eller ryggrad med betydelig begrensning av mobilitet, ofte progressiv.